Yong-Hoon+Kim
-
 KAIST Proposes AI Training Method that will Drastically Shorten Time for Complex Quantum Mechanical Calculations
- Professor Yong-Hoon Kim's team from the School of Electrical Engineering succeeded for the first time in accelerating quantum mechanical electronic structure calculations using a convolutional neural network (CNN) model
- Presenting an AI learning principle of quantum mechanical 3D chemical bonding information, the work is expected to accelerate the computer-assisted designing of next-generation materials and devices
The close relationship between AI and high-performance scientific computing can be seen in the fact that both the 2024 Nobel Prizes in Physics and Chemistry were awarded to scientists for their AI-related research contributions in their respective fields of study. KAIST researchers succeeded in dramatically reducing the computation time for highly sophisticated quantum mechanical computer simulations by predicting atomic-level chemical bonding information distributed in 3D space using a novel AI approach.
KAIST (President Kwang-Hyung Lee) announced on the 30th of October that Professor Yong-Hoon Kim's team from the School of Electrical Engineering developed a 3D computer vision artificial neural network-based computation methodology that bypasses the complex algorithms required for atomic-level quantum mechanical calculations traditionally performed using supercomputers to derive the properties of materials.
< Figure 1. Various methodologies are utilized in the simulation of materials and materials, such as quantum mechanical calculations at the nanometer (nm) level, classical mechanical force fields at the scale of tens to hundreds of nanometers, continuum dynamics calculations at the macroscopic scale, and calculations that mix simulations at different scales. These simulations are already playing a key role in a wide range of basic research and application development fields in combination with informatics techniques. Recently, there have been active efforts to introduce machine learning techniques to radically accelerate simulations, but research on introducing machine learning techniques to quantum mechanical electronic structure calculations, which form the basis of high-scale simulations, is still insufficient. >
The quantum mechanical density functional theory (DFT) calculations using supercomputers have become an essential and standard tool in a wide range of research and development fields, including advanced materials and drug design, as they allow fast and accurate prediction of material properties.
*Density functional theory (DFT): A representative theory of ab initio (first principles) calculations that calculate quantum mechanical properties from the atomic level.
However, practical DFT calculations require generating 3D electron density and solving quantum mechanical equations through a complex, iterative self-consistent field (SCF)* process that must be repeated tens to hundreds of times. This restricts its application to systems with only a few hundred to a few thousand atoms.
*Self-consistent field (SCF): A scientific computing method widely used to solve complex many-body problems that must be described by a number of interconnected simultaneous differential equations.
Professor Yong-Hoon Kim’s research team questioned whether recent advancements in AI techniques could be used to bypass the SCF process. As a result, they developed the DeepSCF model, which accelerates calculations by learning chemical bonding information distributed in a 3D space using neural network algorithms from the field of computer vision.
< Figure 2. The deepSCF methodology developed in this study provides a way to rapidly accelerate DFT calculations by avoiding the self-consistent field process (orange box) that had to be performed repeatedly in traditional quantum mechanical electronic structure calculations through artificial neural network techniques (green box). The self-consistent field process is a process of predicting the 3D electron density, constructing the corresponding potential, and then solving the quantum mechanical Cohn-Sham equations, repeating tens to hundreds of times. The core idea of the deepSCF methodology is that the residual electron density (δρ), which is the difference between the electron density (ρ) and the sum of the electron densities of the constituent atoms (ρ0), corresponds to chemical bonding information, so the self-consistent field process is replaced with a 3D convolutional neural network model. >
The research team focused on the fact that, according to density functional theory, electron density contains all quantum mechanical information of electrons, and that the residual electron density — the difference between the total electron density and the sum of the electron densities of the constituent atoms — contains chemical bonding information. They used this as the target for machine learning.
They then adopted a dataset of organic molecules with various chemical bonding characteristics, and applied random rotations and deformations to the atomic structures of these molecules to further enhance the model’s accuracy and generalization capabilities. Ultimately, the research team demonstrated the validity and efficiency of the DeepSCF methodology on large, complex systems.
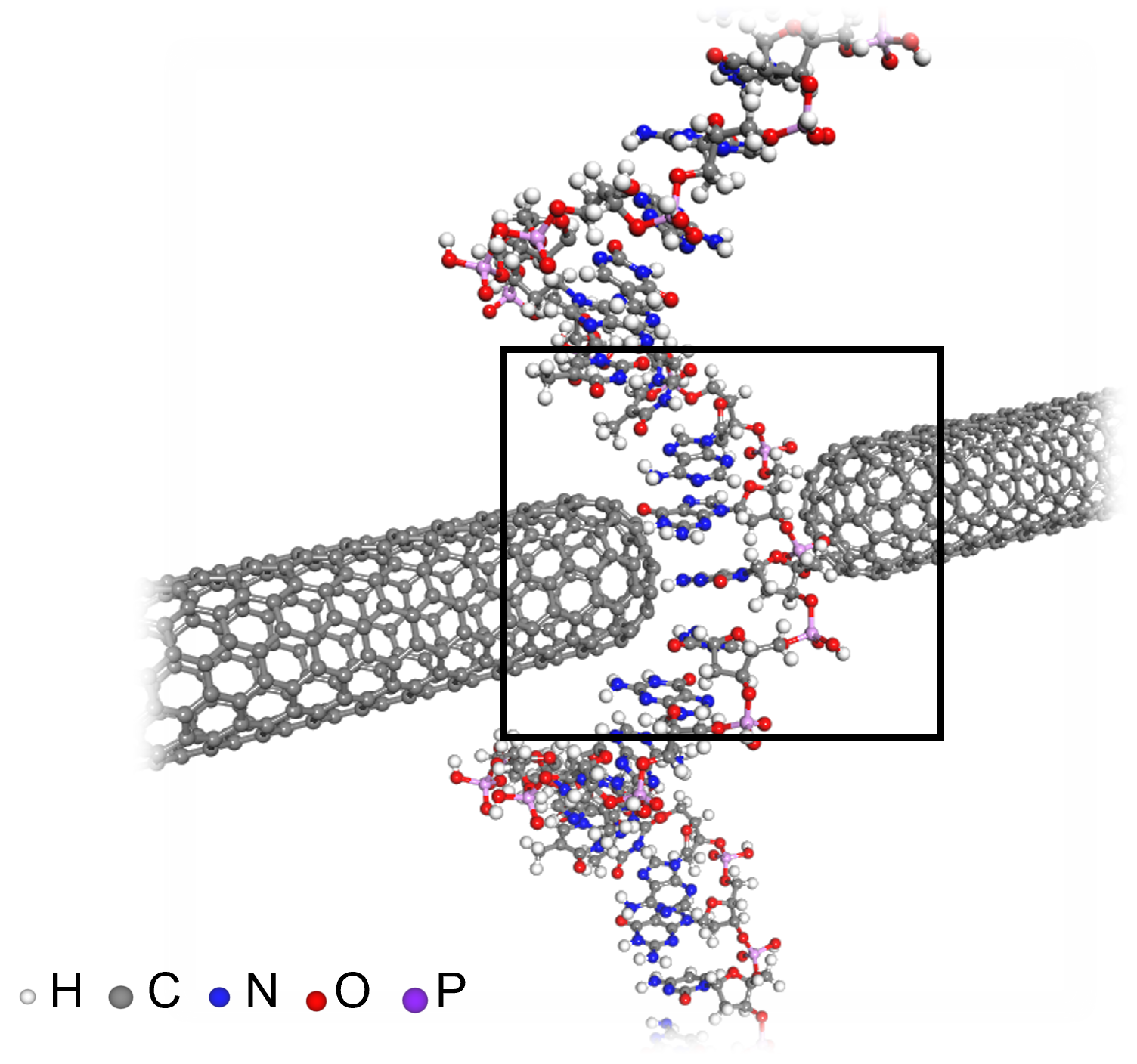
< Figure 3. An example of applying the deepSCF methodology to a carbon nanotube-based DNA sequence analysis device model (top left). In addition to classical mechanical interatomic forces (bottom right), the residual electron density (top right) and quantum mechanical electronic structure properties such as the electronic density of states (DOS) (bottom left) containing information on chemical bonding are rapidly predicted with an accuracy corresponding to the standard DFT calculation results that perform the SCF process. >
Professor Yong-Hoon Kim, who supervised the research, explained that his team had found a way to map quantum mechanical chemical bonding information in a 3D space onto artificial neural networks. He noted, “Since quantum mechanical electron structure calculations underpin materials simulations across all scales, this research establishes a foundational principle for accelerating material calculations using artificial intelligence.”
Ryong-Gyu Lee, a PhD candidate in the School of Electrical Engineering, served as the first author of this research, which was published online on October 24 in Npj Computational Materials, a prestigious journal in the field of material computation. (Paper title: “Convolutional network learning of self-consistent electron density via grid-projected atomic fingerprints”)
This research was conducted with support from the KAIST High-Risk Research Program for Graduate Students and the National Research Foundation of Korea’s Mid-career Researcher Support Program.
2024.10.30 View 7037
KAIST Proposes AI Training Method that will Drastically Shorten Time for Complex Quantum Mechanical Calculations
- Professor Yong-Hoon Kim's team from the School of Electrical Engineering succeeded for the first time in accelerating quantum mechanical electronic structure calculations using a convolutional neural network (CNN) model
- Presenting an AI learning principle of quantum mechanical 3D chemical bonding information, the work is expected to accelerate the computer-assisted designing of next-generation materials and devices
The close relationship between AI and high-performance scientific computing can be seen in the fact that both the 2024 Nobel Prizes in Physics and Chemistry were awarded to scientists for their AI-related research contributions in their respective fields of study. KAIST researchers succeeded in dramatically reducing the computation time for highly sophisticated quantum mechanical computer simulations by predicting atomic-level chemical bonding information distributed in 3D space using a novel AI approach.
KAIST (President Kwang-Hyung Lee) announced on the 30th of October that Professor Yong-Hoon Kim's team from the School of Electrical Engineering developed a 3D computer vision artificial neural network-based computation methodology that bypasses the complex algorithms required for atomic-level quantum mechanical calculations traditionally performed using supercomputers to derive the properties of materials.
< Figure 1. Various methodologies are utilized in the simulation of materials and materials, such as quantum mechanical calculations at the nanometer (nm) level, classical mechanical force fields at the scale of tens to hundreds of nanometers, continuum dynamics calculations at the macroscopic scale, and calculations that mix simulations at different scales. These simulations are already playing a key role in a wide range of basic research and application development fields in combination with informatics techniques. Recently, there have been active efforts to introduce machine learning techniques to radically accelerate simulations, but research on introducing machine learning techniques to quantum mechanical electronic structure calculations, which form the basis of high-scale simulations, is still insufficient. >
The quantum mechanical density functional theory (DFT) calculations using supercomputers have become an essential and standard tool in a wide range of research and development fields, including advanced materials and drug design, as they allow fast and accurate prediction of material properties.
*Density functional theory (DFT): A representative theory of ab initio (first principles) calculations that calculate quantum mechanical properties from the atomic level.
However, practical DFT calculations require generating 3D electron density and solving quantum mechanical equations through a complex, iterative self-consistent field (SCF)* process that must be repeated tens to hundreds of times. This restricts its application to systems with only a few hundred to a few thousand atoms.
*Self-consistent field (SCF): A scientific computing method widely used to solve complex many-body problems that must be described by a number of interconnected simultaneous differential equations.
Professor Yong-Hoon Kim’s research team questioned whether recent advancements in AI techniques could be used to bypass the SCF process. As a result, they developed the DeepSCF model, which accelerates calculations by learning chemical bonding information distributed in a 3D space using neural network algorithms from the field of computer vision.
< Figure 2. The deepSCF methodology developed in this study provides a way to rapidly accelerate DFT calculations by avoiding the self-consistent field process (orange box) that had to be performed repeatedly in traditional quantum mechanical electronic structure calculations through artificial neural network techniques (green box). The self-consistent field process is a process of predicting the 3D electron density, constructing the corresponding potential, and then solving the quantum mechanical Cohn-Sham equations, repeating tens to hundreds of times. The core idea of the deepSCF methodology is that the residual electron density (δρ), which is the difference between the electron density (ρ) and the sum of the electron densities of the constituent atoms (ρ0), corresponds to chemical bonding information, so the self-consistent field process is replaced with a 3D convolutional neural network model. >
The research team focused on the fact that, according to density functional theory, electron density contains all quantum mechanical information of electrons, and that the residual electron density — the difference between the total electron density and the sum of the electron densities of the constituent atoms — contains chemical bonding information. They used this as the target for machine learning.
They then adopted a dataset of organic molecules with various chemical bonding characteristics, and applied random rotations and deformations to the atomic structures of these molecules to further enhance the model’s accuracy and generalization capabilities. Ultimately, the research team demonstrated the validity and efficiency of the DeepSCF methodology on large, complex systems.
< Figure 3. An example of applying the deepSCF methodology to a carbon nanotube-based DNA sequence analysis device model (top left). In addition to classical mechanical interatomic forces (bottom right), the residual electron density (top right) and quantum mechanical electronic structure properties such as the electronic density of states (DOS) (bottom left) containing information on chemical bonding are rapidly predicted with an accuracy corresponding to the standard DFT calculation results that perform the SCF process. >
Professor Yong-Hoon Kim, who supervised the research, explained that his team had found a way to map quantum mechanical chemical bonding information in a 3D space onto artificial neural networks. He noted, “Since quantum mechanical electron structure calculations underpin materials simulations across all scales, this research establishes a foundational principle for accelerating material calculations using artificial intelligence.”
Ryong-Gyu Lee, a PhD candidate in the School of Electrical Engineering, served as the first author of this research, which was published online on October 24 in Npj Computational Materials, a prestigious journal in the field of material computation. (Paper title: “Convolutional network learning of self-consistent electron density via grid-projected atomic fingerprints”)
This research was conducted with support from the KAIST High-Risk Research Program for Graduate Students and the National Research Foundation of Korea’s Mid-career Researcher Support Program.
2024.10.30 View 7037 -
 A Theoretical Boost to Nano-Scale Devices
- Researchers calculate the quasi-Fermi levels in molecular junctions applying an initio approach. -
Semiconductor companies are struggling to develop devices that are mere nanometers in size, and much of the challenge lies in being able to more accurately describe the underlying physics at that nano-scale. But a new computational approach that has been in the works for a decade could break down these barriers.
Devices using semiconductors, from computers to solar cells, have enjoyed tremendous efficiency improvements in the last few decades. Famously, one of the co-founders of Intel, Gordon Moore, observed that the number of transistors in an integrated circuit doubles about every two years—and this ‘Moore’s law’ held true for some time.
In recent years, however, such gains have slowed as firms that attempt to engineer nano-scale transistors hit the limits of miniaturization at the atomic level.
Researchers with the School of Electrical Engineering at KAIST have developed a new approach to the underlying physics of semiconductors.
“With open quantum systems as the main research target of our lab, we were revisiting concepts that had been taken for granted and even appear in standard semiconductor physics textbooks such as the voltage drop in operating semiconductor devices,” said the lead researcher Professor Yong-Hoon Kim. “Questioning how all these concepts could be understood and possibly revised at the nano-scale, it was clear that there was something incomplete about our current understanding.”
“And as the semiconductor chips are being scaled down to the atomic level, coming up with a better theory to describe semiconductor devices has become an urgent task.”
The current understanding states that semiconductors are materials that act like half-way houses between conductors, like copper or steel, and insulators, like rubber or Styrofoam. They sometimes conduct electricity, but not always. This makes them a great material for intentionally controlling the flow of current, which in turn is useful for constructing the simple on/off switches—transistors—that are the foundation of memory and logic devices in computers.
In order to ‘switch on’ a semiconductor, a current or light source is applied, exciting an electron in an atom to jump from what is called a ‘valence band,’ which is filled with electrons, up to the ‘conduction band,’ which is originally unfilled or only partially filled with electrons. Electrons that have jumped up to the conduction band thanks to external stimuli and the remaining ‘holes’ are now able to move about and act as charge carriers to flow electric current.
The physical concept that describes the populations of the electrons in the conduction band and the holes in the valence band and the energy required to make this jump is formulated in terms of the so-called ‘Fermi level.’ For example, you need to know the Fermi levels of the electrons and holes in order to know what amount of energy you are going to get out of a solar cell, including losses.
But the Fermi level concept is only straightforwardly defined so long as a semiconductor device is at equilibrium—sitting on a shelf doing nothing—and the whole point of semiconductor devices is not to leave them on the shelf.
Some 70 years ago, William Shockley, the Nobel Prize-winning co-inventor of the transistor at the Bell Labs, came up with a bit of a theoretical fudge, the ‘quasi-Fermi level,’ or QFL, enabling rough prediction and measurement of the interaction between valence band holes and conduction band electrons, and this has worked pretty well until now.
“But when you are working at the scale of just a few nanometers, the methods to theoretically calculate or experimentally measure the splitting of QFLs were just not available,” said Professor Kim.
This means that at this scale, issues such as errors relating to voltage drop take on much greater significance.
Kim’s team worked for nearly ten years on developing a novel theoretical description of nano-scale quantum electron transport that can replace the standard method—and the software that allows them to put it to use. This involved the further development of a bit of math known as the Density Functional Theory that simplifies the equations describing the interactions of electrons, and which has been very useful in other fields such as high-throughput computational materials discovery.
For the first time, they were able to calculate the QFL splitting, offering a new understanding of the relationship between voltage drop and quantum electron transport in atomic scale devices.
In addition to looking into various interesting non-equilibrium quantum phenomena with their novel methodology, the team is now further developing their software into a computer-aided design tool to be used by semiconductor companies for developing and fabricating advanced semiconductor devices.
The study, featured at the Proceedings of the National Academy of Sciences of the USA on May 12, was supported by the National Research Foundation and the Korea Institute of Science and Technology Information Supercomputing Center.
Image caption: The newly developed formalism and QFL splitting analysis led to new ways of characterizing extremely scaled-down semiconductor devices and the technology computer-aided design (TCAD) of next- generation nano-electronic/energy/bio devices.
Image credit: Yong-Hoon Kim, KAIST
Image usage restrictions: News organizations may use or redistribute this image, with proper attribution, as part of news coverage of this paper only.
Publication:
Juho Lee, Hyeonwoo Yeo, and Yong-Hoon Kim. (2020) ‘Quasi-Fermi level splitting in nanoscale junctions from ab initio.’ Proceedings of the National Academy of Sciences of the United States of America (PNAS), Volume 117, Issue 19, pp.10142-101488. Available online at https://doi.org/10.1073/pnas.1921273117
Profile:
Yong-Hoon Kim
Professor
y.h.kim@kaist.ac.kr
http://nanocore.kaist.ac.kr/
1st-Principles Nano-Device Computing Lab
School of Electrical Engineering
KAIST
(END)
2020.05.15 View 10035
A Theoretical Boost to Nano-Scale Devices
- Researchers calculate the quasi-Fermi levels in molecular junctions applying an initio approach. -
Semiconductor companies are struggling to develop devices that are mere nanometers in size, and much of the challenge lies in being able to more accurately describe the underlying physics at that nano-scale. But a new computational approach that has been in the works for a decade could break down these barriers.
Devices using semiconductors, from computers to solar cells, have enjoyed tremendous efficiency improvements in the last few decades. Famously, one of the co-founders of Intel, Gordon Moore, observed that the number of transistors in an integrated circuit doubles about every two years—and this ‘Moore’s law’ held true for some time.
In recent years, however, such gains have slowed as firms that attempt to engineer nano-scale transistors hit the limits of miniaturization at the atomic level.
Researchers with the School of Electrical Engineering at KAIST have developed a new approach to the underlying physics of semiconductors.
“With open quantum systems as the main research target of our lab, we were revisiting concepts that had been taken for granted and even appear in standard semiconductor physics textbooks such as the voltage drop in operating semiconductor devices,” said the lead researcher Professor Yong-Hoon Kim. “Questioning how all these concepts could be understood and possibly revised at the nano-scale, it was clear that there was something incomplete about our current understanding.”
“And as the semiconductor chips are being scaled down to the atomic level, coming up with a better theory to describe semiconductor devices has become an urgent task.”
The current understanding states that semiconductors are materials that act like half-way houses between conductors, like copper or steel, and insulators, like rubber or Styrofoam. They sometimes conduct electricity, but not always. This makes them a great material for intentionally controlling the flow of current, which in turn is useful for constructing the simple on/off switches—transistors—that are the foundation of memory and logic devices in computers.
In order to ‘switch on’ a semiconductor, a current or light source is applied, exciting an electron in an atom to jump from what is called a ‘valence band,’ which is filled with electrons, up to the ‘conduction band,’ which is originally unfilled or only partially filled with electrons. Electrons that have jumped up to the conduction band thanks to external stimuli and the remaining ‘holes’ are now able to move about and act as charge carriers to flow electric current.
The physical concept that describes the populations of the electrons in the conduction band and the holes in the valence band and the energy required to make this jump is formulated in terms of the so-called ‘Fermi level.’ For example, you need to know the Fermi levels of the electrons and holes in order to know what amount of energy you are going to get out of a solar cell, including losses.
But the Fermi level concept is only straightforwardly defined so long as a semiconductor device is at equilibrium—sitting on a shelf doing nothing—and the whole point of semiconductor devices is not to leave them on the shelf.
Some 70 years ago, William Shockley, the Nobel Prize-winning co-inventor of the transistor at the Bell Labs, came up with a bit of a theoretical fudge, the ‘quasi-Fermi level,’ or QFL, enabling rough prediction and measurement of the interaction between valence band holes and conduction band electrons, and this has worked pretty well until now.
“But when you are working at the scale of just a few nanometers, the methods to theoretically calculate or experimentally measure the splitting of QFLs were just not available,” said Professor Kim.
This means that at this scale, issues such as errors relating to voltage drop take on much greater significance.
Kim’s team worked for nearly ten years on developing a novel theoretical description of nano-scale quantum electron transport that can replace the standard method—and the software that allows them to put it to use. This involved the further development of a bit of math known as the Density Functional Theory that simplifies the equations describing the interactions of electrons, and which has been very useful in other fields such as high-throughput computational materials discovery.
For the first time, they were able to calculate the QFL splitting, offering a new understanding of the relationship between voltage drop and quantum electron transport in atomic scale devices.
In addition to looking into various interesting non-equilibrium quantum phenomena with their novel methodology, the team is now further developing their software into a computer-aided design tool to be used by semiconductor companies for developing and fabricating advanced semiconductor devices.
The study, featured at the Proceedings of the National Academy of Sciences of the USA on May 12, was supported by the National Research Foundation and the Korea Institute of Science and Technology Information Supercomputing Center.
Image caption: The newly developed formalism and QFL splitting analysis led to new ways of characterizing extremely scaled-down semiconductor devices and the technology computer-aided design (TCAD) of next- generation nano-electronic/energy/bio devices.
Image credit: Yong-Hoon Kim, KAIST
Image usage restrictions: News organizations may use or redistribute this image, with proper attribution, as part of news coverage of this paper only.
Publication:
Juho Lee, Hyeonwoo Yeo, and Yong-Hoon Kim. (2020) ‘Quasi-Fermi level splitting in nanoscale junctions from ab initio.’ Proceedings of the National Academy of Sciences of the United States of America (PNAS), Volume 117, Issue 19, pp.10142-101488. Available online at https://doi.org/10.1073/pnas.1921273117
Profile:
Yong-Hoon Kim
Professor
y.h.kim@kaist.ac.kr
http://nanocore.kaist.ac.kr/
1st-Principles Nano-Device Computing Lab
School of Electrical Engineering
KAIST
(END)
2020.05.15 View 10035