Jeong+Ho+Lee
-
 Accurate Detection of Low-Level Somatic Mutation in Intractable Epilepsy
KAIST medical scientists have developed an advanced method for perfectly detecting low-level somatic mutation in patients with intractable epilepsy. Their study showed that deep sequencing replicates of major focal epilepsy genes accurately and efficiently identified low-level somatic mutations in intractable epilepsy.
According to the study, their diagnostic method could increase the accuracy up to 100%, unlike the conventional sequencing analysis, which stands at about 30% accuracy. This work was published in Acta Neuropathologica.
Epilepsy is a neurological disorder common in children. Approximately one third of child patients are diagnosed with intractable epilepsy despite adequate anti-epileptic medication treatment.
Somatic mutations in mTOR pathway genes, SLC35A2, and BRAF are the major genetic causes of intractable epilepsies. A clinical trial to target Focal Cortical Dysplasia type II (FCDII), the mTOR inhibitor is underway at Severance Hospital, their collaborator in Seoul, Korea. However, it is difficult to detect such somatic mutations causing intractable epilepsy because their mutational burden is less than 5%, which is similar to the level of sequencing artifacts. In the clinical field, this has remained a standing challenge for the genetic diagnosis of somatic mutations in intractable epilepsy.
Professor Jeong Ho Lee’s team at the Graduate School of Medical Science and Engineering analyzed paired brain and peripheral tissues from 232 intractable epilepsy patients with various brain pathologies at Severance Hospital using deep sequencing and extracted the major focal epilepsy genes.
They narrowed down target genes to eight major focal epilepsy genes, eliminating almost all of the false positive calls using deep targeted sequencing. As a result, the advanced method robustly increased the accuracy and enabled them to detect low-level somatic mutations in unmatched Formalin Fixed Paraffin Embedded (FFPE) brain samples, the most clinically relevant samples.
Professor Lee conducted this study in collaboration with Professor Dong Suk Kim and Hoon-Chul Kang at Severance Hospital of Yonsei University. He said, “This advanced method of genetic analysis will improve overall patient care by providing more comprehensive genetic counseling and informing decisions on alternative treatments.”
Professor Lee has investigated low-level somatic mutations arising in the brain for a decade. He is developing innovative diagnostics and therapeutics for untreatable brain disorders including intractable epilepsy and glioblastoma at a tech-startup called SoVarGen. “All of the technologies we used during the research were transferred to the company. This research gave us very good momentum to reach the next phase of our startup,” he remarked.
The work was supported by grants from the Suh Kyungbae Foundation, a National Research Foundation of Korea grant funded by the Ministry of Science and ICT, the Korean Health Technology R&D Project from the Ministry of Health & Welfare, and the Netherlands Organization for Health Research and Development.
(Figure: Landscape of somatic and germline mutations identified in intractable epilepsy patients. a Signaling pathways for all of the mutated genes identified in this study. Bold: somatic mutation, Regular: germline mutation. b The distribution of variant allelic frequencies (VAFs) of identified somatic mutations. c The detecting rate and types of identified mutations according to histopathology. Yellow: somatic mutations, green: two-hit mutations, grey: germline mutations.)
2019.08.14 View 30314
Accurate Detection of Low-Level Somatic Mutation in Intractable Epilepsy
KAIST medical scientists have developed an advanced method for perfectly detecting low-level somatic mutation in patients with intractable epilepsy. Their study showed that deep sequencing replicates of major focal epilepsy genes accurately and efficiently identified low-level somatic mutations in intractable epilepsy.
According to the study, their diagnostic method could increase the accuracy up to 100%, unlike the conventional sequencing analysis, which stands at about 30% accuracy. This work was published in Acta Neuropathologica.
Epilepsy is a neurological disorder common in children. Approximately one third of child patients are diagnosed with intractable epilepsy despite adequate anti-epileptic medication treatment.
Somatic mutations in mTOR pathway genes, SLC35A2, and BRAF are the major genetic causes of intractable epilepsies. A clinical trial to target Focal Cortical Dysplasia type II (FCDII), the mTOR inhibitor is underway at Severance Hospital, their collaborator in Seoul, Korea. However, it is difficult to detect such somatic mutations causing intractable epilepsy because their mutational burden is less than 5%, which is similar to the level of sequencing artifacts. In the clinical field, this has remained a standing challenge for the genetic diagnosis of somatic mutations in intractable epilepsy.
Professor Jeong Ho Lee’s team at the Graduate School of Medical Science and Engineering analyzed paired brain and peripheral tissues from 232 intractable epilepsy patients with various brain pathologies at Severance Hospital using deep sequencing and extracted the major focal epilepsy genes.
They narrowed down target genes to eight major focal epilepsy genes, eliminating almost all of the false positive calls using deep targeted sequencing. As a result, the advanced method robustly increased the accuracy and enabled them to detect low-level somatic mutations in unmatched Formalin Fixed Paraffin Embedded (FFPE) brain samples, the most clinically relevant samples.
Professor Lee conducted this study in collaboration with Professor Dong Suk Kim and Hoon-Chul Kang at Severance Hospital of Yonsei University. He said, “This advanced method of genetic analysis will improve overall patient care by providing more comprehensive genetic counseling and informing decisions on alternative treatments.”
Professor Lee has investigated low-level somatic mutations arising in the brain for a decade. He is developing innovative diagnostics and therapeutics for untreatable brain disorders including intractable epilepsy and glioblastoma at a tech-startup called SoVarGen. “All of the technologies we used during the research were transferred to the company. This research gave us very good momentum to reach the next phase of our startup,” he remarked.
The work was supported by grants from the Suh Kyungbae Foundation, a National Research Foundation of Korea grant funded by the Ministry of Science and ICT, the Korean Health Technology R&D Project from the Ministry of Health & Welfare, and the Netherlands Organization for Health Research and Development.
(Figure: Landscape of somatic and germline mutations identified in intractable epilepsy patients. a Signaling pathways for all of the mutated genes identified in this study. Bold: somatic mutation, Regular: germline mutation. b The distribution of variant allelic frequencies (VAFs) of identified somatic mutations. c The detecting rate and types of identified mutations according to histopathology. Yellow: somatic mutations, green: two-hit mutations, grey: germline mutations.)
2019.08.14 View 30314 -
 Deciphering Brain Somatic Mutations Associated with Alzheimer's Disease
Researchers have found a potential link between non-inherited somatic mutations in the brain and the progression of Alzheimer’s disease
Researchers have identified somatic mutations in the brain that could contribute to the development of Alzheimer’s disease (AD). Their findings were published in the journal Nature Communications last week.
Decades worth of research has identified inherited mutations that lead to early-onset familial AD. Inherited mutations, however, are behind at most half the cases of late onset sporadic AD, in which there is no family history of the disease. But the genetic factors causing the other half of these sporadic cases have been unclear.
Professor Jeong Ho Lee at the Graduate School of Medical Science and Engineering and colleagues analysed the DNA present in post-mortem hippocampal formations and in blood samples from people aged 70 to 96 with AD and age-matched controls. They specifically looked for non-inherited somatic mutations in their brains using high-depth whole exome sequencing.
The team developed a bioinformatics pipeline that enabled them to detect low-level brain somatic single nucleotide variations (SNVs) – mutations that involve the substitution of a single nucleotide with another nucleotide. Brain somatic SNVs have been reported on and accumulate throughout our lives and can sometimes be associated with a range of neurological diseases.
The number of somatic SNVs did not differ between individuals with AD and non-demented controls. Interestingly, somatic SNVs in AD brains arise about 4.8 times more slowly than in blood. When the team performed gene-set enrichment tests, 26.9 percent of the AD brain samples had pathogenic brain somatic SNVs known to be linked to hyperphosphorylation of tau proteins, which is one of major hallmarks of AD.
Then, they pinpointed a pathogenic SNV in the PIN1 gene, a cis/trans isomerase that balances phosphorylation in tau proteins, found in one AD patient’s brain. They found the mutation was 4.9 time more abundant in AT8-positive – a marker for hyper-phosphorylated tau proteins– neurons in the entorhinal cortex than the bulk hippocampal tissue. Furthermore, in a series of functional assays, they observed the mutation causing a loss of function in PIN1 and such haploinsufficiency increased the phosphorylation and aggregation of tau proteins.
“Our study provides new insights into the molecular genetic factors behind Alzheimer’s disease and other neurodegenerative diseases potentially linked to somatic mutations in the brain,” said Professor Lee.
The team is planning to expand their study to a larger cohort in order to establish stronger links between these brain somatic mutations and the pathogenesis of Alzheimer’s disease.
(Figure 1. Bioinformatic pipeline for detecting low-level brain somatic mutations in AD and non-AD.)
(Figure 2. Pathogenic brain somatic mutations associated with tau phosphorylation are significantly enriched in AD brains.)
(Figure 3. A pathogenic brain somatic mutation in PIN1 (c. 477 C>T) is a loss-of-function and related functional assays show its haploinsufficiency increases phosphorylation and aggregation of tau.)
2019.07.19 View 37357
Deciphering Brain Somatic Mutations Associated with Alzheimer's Disease
Researchers have found a potential link between non-inherited somatic mutations in the brain and the progression of Alzheimer’s disease
Researchers have identified somatic mutations in the brain that could contribute to the development of Alzheimer’s disease (AD). Their findings were published in the journal Nature Communications last week.
Decades worth of research has identified inherited mutations that lead to early-onset familial AD. Inherited mutations, however, are behind at most half the cases of late onset sporadic AD, in which there is no family history of the disease. But the genetic factors causing the other half of these sporadic cases have been unclear.
Professor Jeong Ho Lee at the Graduate School of Medical Science and Engineering and colleagues analysed the DNA present in post-mortem hippocampal formations and in blood samples from people aged 70 to 96 with AD and age-matched controls. They specifically looked for non-inherited somatic mutations in their brains using high-depth whole exome sequencing.
The team developed a bioinformatics pipeline that enabled them to detect low-level brain somatic single nucleotide variations (SNVs) – mutations that involve the substitution of a single nucleotide with another nucleotide. Brain somatic SNVs have been reported on and accumulate throughout our lives and can sometimes be associated with a range of neurological diseases.
The number of somatic SNVs did not differ between individuals with AD and non-demented controls. Interestingly, somatic SNVs in AD brains arise about 4.8 times more slowly than in blood. When the team performed gene-set enrichment tests, 26.9 percent of the AD brain samples had pathogenic brain somatic SNVs known to be linked to hyperphosphorylation of tau proteins, which is one of major hallmarks of AD.
Then, they pinpointed a pathogenic SNV in the PIN1 gene, a cis/trans isomerase that balances phosphorylation in tau proteins, found in one AD patient’s brain. They found the mutation was 4.9 time more abundant in AT8-positive – a marker for hyper-phosphorylated tau proteins– neurons in the entorhinal cortex than the bulk hippocampal tissue. Furthermore, in a series of functional assays, they observed the mutation causing a loss of function in PIN1 and such haploinsufficiency increased the phosphorylation and aggregation of tau proteins.
“Our study provides new insights into the molecular genetic factors behind Alzheimer’s disease and other neurodegenerative diseases potentially linked to somatic mutations in the brain,” said Professor Lee.
The team is planning to expand their study to a larger cohort in order to establish stronger links between these brain somatic mutations and the pathogenesis of Alzheimer’s disease.
(Figure 1. Bioinformatic pipeline for detecting low-level brain somatic mutations in AD and non-AD.)
(Figure 2. Pathogenic brain somatic mutations associated with tau phosphorylation are significantly enriched in AD brains.)
(Figure 3. A pathogenic brain somatic mutation in PIN1 (c. 477 C>T) is a loss-of-function and related functional assays show its haploinsufficiency increases phosphorylation and aggregation of tau.)
2019.07.19 View 37357 -
 Understanding Epilepsy in Pediatric Tumors; New Therapeutic Target of Intractable Epilepsy Identified
Pediatric brain tumors are characterized by frequent complications due to intractable epilepsy compared to adult brain tumors. However, the genetic cause of refractory epilepsy in pediatric brain tumors has not been elucidated yet, and it is difficult to treat patients because the tumors do not respond to existing antiepileptic drugs and debilitate children’s development.
A research team led by Professor Jeong Ho Lee of the Graduate School of Medical Science and Engineering has recently identified a neuronal BRAF somatic mutation that causes intrinsic epileptogenicity in pediatric brain tumors. Their research results were published online in Nature Medicine on September 17.
The research team studied patients’ tissue diagnosed with ganglioglioma (GG), one of the main causes of tumor-associated intractable epilepsy, and found that the BRAF V600E somatic mutation is involved in the development of neural stem cells by using deep DNA sequencing. This mutation was carried out in an animal model to reproduce the pathology of GG and to observe seizures to establish an animal model for the treatment of epileptic seizures caused by pediatric brain tumors.
Using immunohistochemical and transcriptome analysis, they realized that the BRAF V600E mutation that arose in early progenitor cells during embryonic brain formation led to the acquisition of intrinsic epileptogenic properties in neuronal lineage cells, whereas tumorigenic properties were attributed to a high proliferation of glial lineage cells exhibiting the mutation. Notably, researchers found that seizures in mice were significantly alleviated by intraventricular infusion of the BRAF V600E inhibitor, Vemurafenib, a clinical anticancer drug.
The authors said, “Our study offers the first direct evidence that the BRAF somatic mutation arising from neural stem cells plays a key role in epileptogenesis in the brain tumor. This study also showed a new therapeutic target for tumor-associated epileptic disorders.”
In collaboration with the KAIST startup company, SoVarGen, the research team is currently developing innovative therapeutics for epileptic seizures derived from pediatric brain tumors. This study was supported by the Suh Kyungbae Foundation (SUHF) and the Citizens United for Research in Epilepsy.
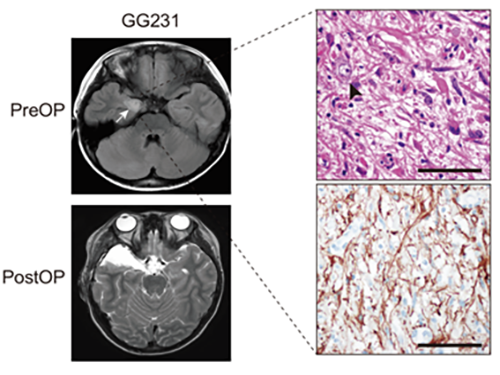
(Figure: Preoperative and postoperative brain MRI (left panel), tumor H&E (right upper panel) and GFAP immunohistochemical (right lower panel) staining images from a patient with ganglioglioma (GG231) carrying the BRAFV600E mutation. The white arrow and the black arrowhead indicate the brain tumor and a dysplastic neuron, respectively.)
2018.09.19 View 6053
Understanding Epilepsy in Pediatric Tumors; New Therapeutic Target of Intractable Epilepsy Identified
Pediatric brain tumors are characterized by frequent complications due to intractable epilepsy compared to adult brain tumors. However, the genetic cause of refractory epilepsy in pediatric brain tumors has not been elucidated yet, and it is difficult to treat patients because the tumors do not respond to existing antiepileptic drugs and debilitate children’s development.
A research team led by Professor Jeong Ho Lee of the Graduate School of Medical Science and Engineering has recently identified a neuronal BRAF somatic mutation that causes intrinsic epileptogenicity in pediatric brain tumors. Their research results were published online in Nature Medicine on September 17.
The research team studied patients’ tissue diagnosed with ganglioglioma (GG), one of the main causes of tumor-associated intractable epilepsy, and found that the BRAF V600E somatic mutation is involved in the development of neural stem cells by using deep DNA sequencing. This mutation was carried out in an animal model to reproduce the pathology of GG and to observe seizures to establish an animal model for the treatment of epileptic seizures caused by pediatric brain tumors.
Using immunohistochemical and transcriptome analysis, they realized that the BRAF V600E mutation that arose in early progenitor cells during embryonic brain formation led to the acquisition of intrinsic epileptogenic properties in neuronal lineage cells, whereas tumorigenic properties were attributed to a high proliferation of glial lineage cells exhibiting the mutation. Notably, researchers found that seizures in mice were significantly alleviated by intraventricular infusion of the BRAF V600E inhibitor, Vemurafenib, a clinical anticancer drug.
The authors said, “Our study offers the first direct evidence that the BRAF somatic mutation arising from neural stem cells plays a key role in epileptogenesis in the brain tumor. This study also showed a new therapeutic target for tumor-associated epileptic disorders.”
In collaboration with the KAIST startup company, SoVarGen, the research team is currently developing innovative therapeutics for epileptic seizures derived from pediatric brain tumors. This study was supported by the Suh Kyungbae Foundation (SUHF) and the Citizens United for Research in Epilepsy.
(Figure: Preoperative and postoperative brain MRI (left panel), tumor H&E (right upper panel) and GFAP immunohistochemical (right lower panel) staining images from a patient with ganglioglioma (GG231) carrying the BRAFV600E mutation. The white arrow and the black arrowhead indicate the brain tumor and a dysplastic neuron, respectively.)
2018.09.19 View 6053 -
 A Breakthrough for Understanding Glioblastoma: Origin Cells for Deadly Brain Tumors Identified
Figure 1. The pattern of GBM genesis is similar to that of firework. The bottom canon represents the first occurrence of the SVZ mutated cell.
A new study by KAIST researchers identified where the mutation causing glioblastoma starts. According to the study, neural stem cells away from the tumor mass are the cells of origin that contain mutation drivers for glioblastoma, one of the most aggressive brain tumor. This breakthrough research, reported in Nature on August 1, gives insights for understanding why glioblastomas almost always grow back, even after surgery, and suggests novel ways to treat glioblastoma, which was previously thought to be incurable.
Like most cancers, glioblastoma is treated with surgery to remove as much of the tumor as possible, then radiation and chemotherapy. However, it almost always returns in less than a year and its median survival time is only 15 months. Precision therapeutic approaches targeting tumors themselves didn’t lead to any breakthroughs.
Professor Jeong Ho Lee’s team at the Graduate School of Medical Science and Engineering described direct genetic evidence through the deep sequencing of all triple-matched samples: normal SVZ tissue away from the tumor mass, tumor tissue, and normal cortical tissue. The research team studied 28 patients with glioblastomas and other types of brain tumors who underwent supra-total resection or other surgical resections of tumors, providing access to normal subventricular zone (SVZ) tissue (where neural stem cells are located) away from the tumor mass. The researchers used various deep and single cell sequencing technologies to conduct comparative DNA analysis on the samples from the patient’s SVZ tissue and tumors.
They reported that normal SVZ tissue away from the tumor in 56.3% of patients with glioblastoma already contained low-level glioblastoma driver mutations that were observed at high levels in their matching tumors. Furthermore, the research team generated a genome edited mouse carrying glioblastoma mutations in the SVZ and showed that neural stem cells with mutations migrate from the SVZ lead to the development of glioblastomas in distant brain regions. (See the image below)
Professor Lee conducted this study in collaboration with Professor Seok-Gu Kang of the Brain Tumor Center at Severance Hospital of Yonsei University. He said, “It’s easier to understand when we compare it to fireworks. Every flare flying around sky can be likened to cancer cells even though the fireworks are triggered on the ground. We found the trigger.” The identification of this mutation pathway of glioblastomas will lead to a new paradigm for therapeutic strategies. He added, “Now, we can focus on interrupting the recurrence and evolution of glioblastomas.”
Professor Lee has investigated mutations arising in the brain for a decade. He is developing innovative diagnostics and therapeutics for untreatable brain disorders including intractable epilepsy and glioblastoma at a tech-startup, SoVarGen. “All technologies we used during the research were transferred to the company. This research gave us very good momentum to reach the next phase of our startup,” he remarked.
Figure 2. Genetic analysis of tumor-free SVZ tissue and matching tumor tissue from GBM patients.
Figure 3. Glioma progression in genome edited mice carrying GBM mutations in the SVZ
2018.08.02 View 12905
A Breakthrough for Understanding Glioblastoma: Origin Cells for Deadly Brain Tumors Identified
Figure 1. The pattern of GBM genesis is similar to that of firework. The bottom canon represents the first occurrence of the SVZ mutated cell.
A new study by KAIST researchers identified where the mutation causing glioblastoma starts. According to the study, neural stem cells away from the tumor mass are the cells of origin that contain mutation drivers for glioblastoma, one of the most aggressive brain tumor. This breakthrough research, reported in Nature on August 1, gives insights for understanding why glioblastomas almost always grow back, even after surgery, and suggests novel ways to treat glioblastoma, which was previously thought to be incurable.
Like most cancers, glioblastoma is treated with surgery to remove as much of the tumor as possible, then radiation and chemotherapy. However, it almost always returns in less than a year and its median survival time is only 15 months. Precision therapeutic approaches targeting tumors themselves didn’t lead to any breakthroughs.
Professor Jeong Ho Lee’s team at the Graduate School of Medical Science and Engineering described direct genetic evidence through the deep sequencing of all triple-matched samples: normal SVZ tissue away from the tumor mass, tumor tissue, and normal cortical tissue. The research team studied 28 patients with glioblastomas and other types of brain tumors who underwent supra-total resection or other surgical resections of tumors, providing access to normal subventricular zone (SVZ) tissue (where neural stem cells are located) away from the tumor mass. The researchers used various deep and single cell sequencing technologies to conduct comparative DNA analysis on the samples from the patient’s SVZ tissue and tumors.
They reported that normal SVZ tissue away from the tumor in 56.3% of patients with glioblastoma already contained low-level glioblastoma driver mutations that were observed at high levels in their matching tumors. Furthermore, the research team generated a genome edited mouse carrying glioblastoma mutations in the SVZ and showed that neural stem cells with mutations migrate from the SVZ lead to the development of glioblastomas in distant brain regions. (See the image below)
Professor Lee conducted this study in collaboration with Professor Seok-Gu Kang of the Brain Tumor Center at Severance Hospital of Yonsei University. He said, “It’s easier to understand when we compare it to fireworks. Every flare flying around sky can be likened to cancer cells even though the fireworks are triggered on the ground. We found the trigger.” The identification of this mutation pathway of glioblastomas will lead to a new paradigm for therapeutic strategies. He added, “Now, we can focus on interrupting the recurrence and evolution of glioblastomas.”
Professor Lee has investigated mutations arising in the brain for a decade. He is developing innovative diagnostics and therapeutics for untreatable brain disorders including intractable epilepsy and glioblastoma at a tech-startup, SoVarGen. “All technologies we used during the research were transferred to the company. This research gave us very good momentum to reach the next phase of our startup,” he remarked.
Figure 2. Genetic analysis of tumor-free SVZ tissue and matching tumor tissue from GBM patients.
Figure 3. Glioma progression in genome edited mice carrying GBM mutations in the SVZ
2018.08.02 View 12905 -
 Mechanism Leading to Cortical Malformation from Brain-Only Mutations Identified
Focal malformations of cortical development (FMCDs) are a heterogeneous group of brain cortical abnormalities. These conditions are the most common causes of medically refractory epilepsy in children and are highly associated with intellectual disability, developmental delay, and autism-spectrum disorders. Despite a broad spectrum of cortical abnormalities in FMCDs, the defective migration of neuronal cells is considered a key pathological hallmark.
A research team led by Professor Jeong Ho Lee in the Graduate School of Medical Science and Engineering at KAIST has recently investigated the molecular mechanism of defective neuronal migration in FMCDs. Their research results were published online in Neuron on June 21, 2018.
The research team previously demonstrated that brain-only mutations in the mechanistic target of rapamycin (MTOR) gene causes focal cortical dysplasia, one major form of FMCDs leading to intractable epilepsy in children. However, the molecular mechanisms by which brain-only mutations in MTOR lead to cortical dyslamination and defective neuronal migration in FMCDs remain unclear.
To study the molecular mechanism of brain cortical dyslamination, the research team utilized patients’ brain tissues and modeled the MTOR mutation-carrying cell and animal models recapitulating the pathogenesis and symptoms of FMCD patients. By performing comprehensive molecular genetic experiments, they found that the formation of primary cilia, one of cellular organelles, was disrupted in MTOR mutation-carrying neurons and demonstrated that this ciliary disruption was a cause of cortical dyslamination in FMCDs.
MTOR mutations prevented degradation of the OFD1 protein, one of the negative regulators of ciliary formation. As a result, the OFD1 protein was abnormally accumulated in MTOR mutation-carrying neurons, causing focal cortical dyslamination. By suppressing the expression of the OFD1 protein, the research team was able to rescue the defective formation of primary cilia, leading to the restoration of cortical dyslamination and defective neuronal migration considerably.
Based on these results, the research team is carrying out further research to develop novel therapeutics for patients with FMCDs caused by brain-only mutations.
This work was supported by grants from the Suh Kyungbae Foundation and Citizens United for Research in Epilepsy.
The research paper is titled “Brain Somatic Mutations in MTOR Disrupt Neuronal Ciliogenesis, Leading to Focal Cortical Dyslamination.” (Digital Object Identifier #: 10.1016/j.neuron.2018.05.039)
Picture 1: The disrupted formation of primary cilia in brain tissues of FMCD mouse models and patients with FMCDs caused by brain somatic mutations in MTOR.
Picture 2: The rescue of defective ciliary formation in FMCD mouse models leading to the restoration of cortical dyslamination and defective neuronal migration.
2018.07.02 View 8930
Mechanism Leading to Cortical Malformation from Brain-Only Mutations Identified
Focal malformations of cortical development (FMCDs) are a heterogeneous group of brain cortical abnormalities. These conditions are the most common causes of medically refractory epilepsy in children and are highly associated with intellectual disability, developmental delay, and autism-spectrum disorders. Despite a broad spectrum of cortical abnormalities in FMCDs, the defective migration of neuronal cells is considered a key pathological hallmark.
A research team led by Professor Jeong Ho Lee in the Graduate School of Medical Science and Engineering at KAIST has recently investigated the molecular mechanism of defective neuronal migration in FMCDs. Their research results were published online in Neuron on June 21, 2018.
The research team previously demonstrated that brain-only mutations in the mechanistic target of rapamycin (MTOR) gene causes focal cortical dysplasia, one major form of FMCDs leading to intractable epilepsy in children. However, the molecular mechanisms by which brain-only mutations in MTOR lead to cortical dyslamination and defective neuronal migration in FMCDs remain unclear.
To study the molecular mechanism of brain cortical dyslamination, the research team utilized patients’ brain tissues and modeled the MTOR mutation-carrying cell and animal models recapitulating the pathogenesis and symptoms of FMCD patients. By performing comprehensive molecular genetic experiments, they found that the formation of primary cilia, one of cellular organelles, was disrupted in MTOR mutation-carrying neurons and demonstrated that this ciliary disruption was a cause of cortical dyslamination in FMCDs.
MTOR mutations prevented degradation of the OFD1 protein, one of the negative regulators of ciliary formation. As a result, the OFD1 protein was abnormally accumulated in MTOR mutation-carrying neurons, causing focal cortical dyslamination. By suppressing the expression of the OFD1 protein, the research team was able to rescue the defective formation of primary cilia, leading to the restoration of cortical dyslamination and defective neuronal migration considerably.
Based on these results, the research team is carrying out further research to develop novel therapeutics for patients with FMCDs caused by brain-only mutations.
This work was supported by grants from the Suh Kyungbae Foundation and Citizens United for Research in Epilepsy.
The research paper is titled “Brain Somatic Mutations in MTOR Disrupt Neuronal Ciliogenesis, Leading to Focal Cortical Dyslamination.” (Digital Object Identifier #: 10.1016/j.neuron.2018.05.039)
Picture 1: The disrupted formation of primary cilia in brain tissues of FMCD mouse models and patients with FMCDs caused by brain somatic mutations in MTOR.
Picture 2: The rescue of defective ciliary formation in FMCD mouse models leading to the restoration of cortical dyslamination and defective neuronal migration.
2018.07.02 View 8930 -
 Mutations Occurring Only in Brain Responsible for Intractable Epilepsy Identified
KAIST researchers have discovered that brain somatic mutations in MTOR gene induce intractable epilepsy and suggest a precision medicine to treat epileptic seizures.
Epilepsy is a brain disorder which afflicts more than 50 million people worldwide. Many epilepsy patients can control their symptoms through medication, but about 30% suffer from intractable epilepsy and are unable to manage the disease with drugs. Intractable epilepsy causes multiple seizures, permanent mental, physical, and developmental disabilities, and even death. Therefore, surgical removal of the affected area from the brain has been practiced as a treatment for patients with medically refractory seizures, but this too fails to provide a complete solution because only 60% of the patients who undergo surgery are rendered free of seizures.
A Korean research team led by Professor Jeong Ho Lee of the Graduate School of Medical Science and Engineering at the Korea Advanced Institute of Science and Technology (KAIST) and Professor Dong-Seok Kim of Epilepsy Research Center at Yonsei University College of Medicine has recently identified brain somatic mutations in the gene of mechanistic target of rapamycin (MTOR) as the cause of focal cortical dysplasia type II (FCDII), one of the most important and common inducers to intractable epilepsy, particularly in children. They propose a targeted therapy to lessen epileptic seizures by suppressing the activation of mTOR kinase, a signaling protein in the brain. Their research results were published online in Nature Medicine on March 23, 2015.
FCDII contributes to the abnormal developments of the cerebral cortex, ranging from cortical disruption to severe forms of cortical dyslamination, balloon cells, and dysplastic neurons. The research team studied 77 FCDII patients with intractable epilepsy who had received a surgery to remove the affected regions from the brain. The researchers used various deep sequencing technologies to conduct comparative DNA analysis of the samples obtained from the patients’ brain and blood, or saliva. They reported that about 16% of the studied patients had somatic mutations in their brain. Such mutations, however, did not take place in their blood or saliva DNA.
Professor Jeong Ho Lee of KAIST said, “This is an important finding. Unlike our previous belief that genetic mutations causing intractable epilepsy exist anywhere in the human body including blood, specific gene mutations incurred only in the brain can lead to intractable epilepsy. From our animal models, we could see how a small fraction of mutations carrying neurons in the brain could affect its entire function.”
The research team recapitulated the pathogenesis of intractable epilepsy by inducing the focal cortical expression of mutated mTOR in the mouse brain via electroporation method and observed as the mouse develop epileptic symptoms. They then treated these mice with the drug called “rapamycin” to inhibit the activity of mTOR protein and observed that it suppressed the development of epileptic seizures with cytomegalic neurons.
“Our study offers the first evidence that brain-somatic activating mutations in MTOR cause FCDII and identifies mTOR as a treatment target for intractable epilepsy,” said co-author Dr. Dong-Seok Kim, a neurosurgeon at Yonsei Medical Center with the country’s largest surgical experiences in treating patients with this condition.
The research paper is titled “Brain somatic mutations in MTOR cause focal cortical dysplasia type II leading to intractable epilepsy.” (Digital Object Identifier #: 10.1038/nm.3824)
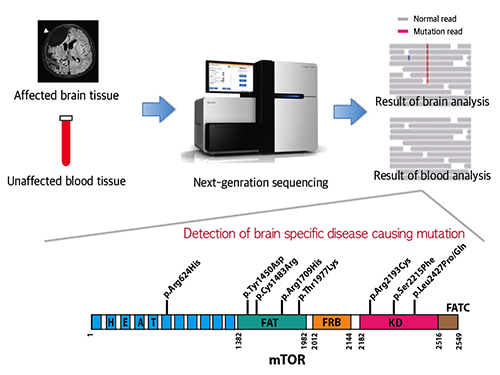
Picture 1: A schematic image to show how to detect brain specific mutation using next-generation sequencing technology with blood-brain paired sample. Simple comparison of non-overlapping mutations between affected and unaffected tissues is able to detect brain specific mutations.
Picture 2: A schematic image to show how to generate focal cortical dysplasia mouse model. This mouse model open the new window of drug screening for seizure patients.
Picture 3: Targeted medicine can rescue the focal cortical dysplasia symptoms including cytomegalic neuron & intractable epilepsy.
2015.03.25 View 15112
Mutations Occurring Only in Brain Responsible for Intractable Epilepsy Identified
KAIST researchers have discovered that brain somatic mutations in MTOR gene induce intractable epilepsy and suggest a precision medicine to treat epileptic seizures.
Epilepsy is a brain disorder which afflicts more than 50 million people worldwide. Many epilepsy patients can control their symptoms through medication, but about 30% suffer from intractable epilepsy and are unable to manage the disease with drugs. Intractable epilepsy causes multiple seizures, permanent mental, physical, and developmental disabilities, and even death. Therefore, surgical removal of the affected area from the brain has been practiced as a treatment for patients with medically refractory seizures, but this too fails to provide a complete solution because only 60% of the patients who undergo surgery are rendered free of seizures.
A Korean research team led by Professor Jeong Ho Lee of the Graduate School of Medical Science and Engineering at the Korea Advanced Institute of Science and Technology (KAIST) and Professor Dong-Seok Kim of Epilepsy Research Center at Yonsei University College of Medicine has recently identified brain somatic mutations in the gene of mechanistic target of rapamycin (MTOR) as the cause of focal cortical dysplasia type II (FCDII), one of the most important and common inducers to intractable epilepsy, particularly in children. They propose a targeted therapy to lessen epileptic seizures by suppressing the activation of mTOR kinase, a signaling protein in the brain. Their research results were published online in Nature Medicine on March 23, 2015.
FCDII contributes to the abnormal developments of the cerebral cortex, ranging from cortical disruption to severe forms of cortical dyslamination, balloon cells, and dysplastic neurons. The research team studied 77 FCDII patients with intractable epilepsy who had received a surgery to remove the affected regions from the brain. The researchers used various deep sequencing technologies to conduct comparative DNA analysis of the samples obtained from the patients’ brain and blood, or saliva. They reported that about 16% of the studied patients had somatic mutations in their brain. Such mutations, however, did not take place in their blood or saliva DNA.
Professor Jeong Ho Lee of KAIST said, “This is an important finding. Unlike our previous belief that genetic mutations causing intractable epilepsy exist anywhere in the human body including blood, specific gene mutations incurred only in the brain can lead to intractable epilepsy. From our animal models, we could see how a small fraction of mutations carrying neurons in the brain could affect its entire function.”
The research team recapitulated the pathogenesis of intractable epilepsy by inducing the focal cortical expression of mutated mTOR in the mouse brain via electroporation method and observed as the mouse develop epileptic symptoms. They then treated these mice with the drug called “rapamycin” to inhibit the activity of mTOR protein and observed that it suppressed the development of epileptic seizures with cytomegalic neurons.
“Our study offers the first evidence that brain-somatic activating mutations in MTOR cause FCDII and identifies mTOR as a treatment target for intractable epilepsy,” said co-author Dr. Dong-Seok Kim, a neurosurgeon at Yonsei Medical Center with the country’s largest surgical experiences in treating patients with this condition.
The research paper is titled “Brain somatic mutations in MTOR cause focal cortical dysplasia type II leading to intractable epilepsy.” (Digital Object Identifier #: 10.1038/nm.3824)
Picture 1: A schematic image to show how to detect brain specific mutation using next-generation sequencing technology with blood-brain paired sample. Simple comparison of non-overlapping mutations between affected and unaffected tissues is able to detect brain specific mutations.
Picture 2: A schematic image to show how to generate focal cortical dysplasia mouse model. This mouse model open the new window of drug screening for seizure patients.
Picture 3: Targeted medicine can rescue the focal cortical dysplasia symptoms including cytomegalic neuron & intractable epilepsy.
2015.03.25 View 15112