Gliabiology+Lab
-
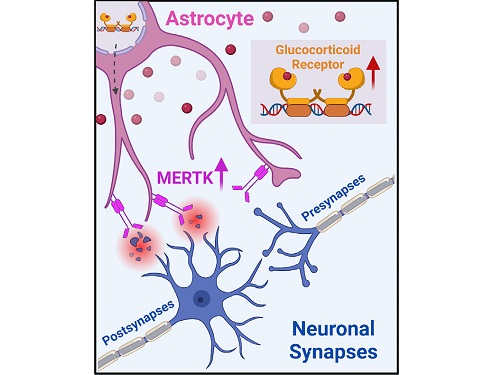 A KAIST research team identifies a cause of mental diseases induced by childhood abuse
Childhood neglect and/or abuse can induce extreme stress that significantly changes neural networks and functions during growth. This can lead to mental illnesses, including depression and schizophrenia, but the exact mechanism and means to control it were yet to be discovered.
On August 1, a KAIST research team led by Professor Won-Suk Chung from the Department of Biological Sciences announced the identification of excessive synapse removal mediated by astrocytes as the cause of mental diseases induced by childhood abuse trauma. Their research was published in Immunity, a top international journal in the field of immunology.
The research team discovered that the excessive astrocyte-mediated removal of excitatory synapses in the brain in response to stress hormones is a cause of mental diseases induced by childhood neglect and abuse. Clinical data have previously shown that high levels of stress can lead to various mental diseases, but the exact mechanism has been unknown. The results of this research therefore are expected to be widely applied to the prevention and treatment of such diseases.
The research team clinically screened an FDA-approved drug to uncover the mechanism that regulates the phagocytotic role of astrocytes, in which they capture external substances and eliminate them. As a result, the team found that synthetic glucocorticoids, namely stress hormones, enhanced astrocyte-mediated phagocytosis to an abnormal level. Glucocorticoids play essential roles in processes that maintain life, such as carbohydrate metabolism and anti-inflammation, but are also secreted in response to external stimuli such as stress, allowing the body to respond appropriately. However, excessive and long-term exposure to glucocorticoids caused by chronic stress can lead to various mental diseases including depression, cognitive disorders, and anxiety.
< Figure 1. Results of screening for compounds that increase astrocyte phagocytosis
(A) Discovered that synthetic glucocorticoid (stress hormone) increases the phagocytosis of astrocytes through screening of FDA-approved clinical compounds. (B-C) When treated with stress hormones, the phagocytosis of astrocytes is greatly increased, but this phenomenon is strongly suppressed by the GR antagonist (Mifepristone). CORT: corticosterone (stress hormone), Eplerenone: mineralocorticoid receptor (MR) antagonist, Mifepristone: glucocorticoid receptor (GR) antagonist >
To understand the changes in astrocyte functions caused by childhood stress, the research team used mice models with early social deprivation, and discovered that stress hormones bind to the glucocorticoid receptors (GRs) of astrocytes. This significantly increased the expression of Mer tyrosine kinase (MERK), which plays an essential role in astrocyte phagocytosis. Surprisingly, out of the various neurons in the cerebral cortex, astrocytes would eliminate only the excitatory synapses of specific neurons. The team found that this builds abnormal neural networks, which can lead to complex behavioral abnormalities such as social deficiencies and depression in adulthood.
The team also observed that microglia, which also play an important role in cerebral immunity, did not contribute to synapse removal in the mice models with early social deprivation. This confirms that the response to stress hormones during childhood is specifically astrocyte-mediated.
To find out whether these results are also applicable in humans, the research team used a brain organoid grown from human-induced pluripotent stem cells to observe human responses to stress hormones. The team observed that the stress hormones induced astrocyte GRs and phagocyte activation in the human brain organoid as well, and confirmed that the astrocytes subsequently eliminated excessive amounts of excitatory synapses. By showing that mice and humans both showed the same synapse control mechanism in response to stress, the team suggested that this discovery is applicable to mental disorders in humans.
< Figure 2. A schematic diagram of the study published in Immunity. Excessive stress hormone secretion in childhood increases the expression of the MERTK phagocytic receptor through the glucocorticoid receptor (GR) of astrocytes, resulting in excessive elimination of excitatory synapses. Excessive synaptic elimination by astrocytes during brain development causes permanent damage to brain circuits, resulting in abnormal neural activity in the adult brain and psychiatric behaviors such as depression and anti-social tendencies. >
Prof. Won-Suk Chung said, “Until now, we did not know the exact mechanism for how childhood stress caused brain diseases. This research was the first to show that the excessive phagocytosis of astrocytes could be an important cause of such diseases.” He added, “In the future, controlling the immune response of astrocytes will be used as a fundamental target for understanding and treating brain diseases.”
This research, written by co-first authors Youkyeong Byun (Ph.D. candidate) and Nam-Shik Kim (post-doctoral associate) from the KAIST Department of Biological Sciences, was published in the internationally renowned journal Immunity, a sister magazine of Cell and one of the best journal in the field of immunology, on July 31 under the title "Stress induces behavioral abnormalities by increasing expression of phagocytic receptor MERTK in astrocytes to promote synapse phagocytosis."
This work was supported by a National Research Foundation of Korea grant, the Korea Health Industry Development Institute (KHIDI), and the Korea Dementia Research Center (KDRC).
2023.08.04 View 8169
A KAIST research team identifies a cause of mental diseases induced by childhood abuse
Childhood neglect and/or abuse can induce extreme stress that significantly changes neural networks and functions during growth. This can lead to mental illnesses, including depression and schizophrenia, but the exact mechanism and means to control it were yet to be discovered.
On August 1, a KAIST research team led by Professor Won-Suk Chung from the Department of Biological Sciences announced the identification of excessive synapse removal mediated by astrocytes as the cause of mental diseases induced by childhood abuse trauma. Their research was published in Immunity, a top international journal in the field of immunology.
The research team discovered that the excessive astrocyte-mediated removal of excitatory synapses in the brain in response to stress hormones is a cause of mental diseases induced by childhood neglect and abuse. Clinical data have previously shown that high levels of stress can lead to various mental diseases, but the exact mechanism has been unknown. The results of this research therefore are expected to be widely applied to the prevention and treatment of such diseases.
The research team clinically screened an FDA-approved drug to uncover the mechanism that regulates the phagocytotic role of astrocytes, in which they capture external substances and eliminate them. As a result, the team found that synthetic glucocorticoids, namely stress hormones, enhanced astrocyte-mediated phagocytosis to an abnormal level. Glucocorticoids play essential roles in processes that maintain life, such as carbohydrate metabolism and anti-inflammation, but are also secreted in response to external stimuli such as stress, allowing the body to respond appropriately. However, excessive and long-term exposure to glucocorticoids caused by chronic stress can lead to various mental diseases including depression, cognitive disorders, and anxiety.
< Figure 1. Results of screening for compounds that increase astrocyte phagocytosis
(A) Discovered that synthetic glucocorticoid (stress hormone) increases the phagocytosis of astrocytes through screening of FDA-approved clinical compounds. (B-C) When treated with stress hormones, the phagocytosis of astrocytes is greatly increased, but this phenomenon is strongly suppressed by the GR antagonist (Mifepristone). CORT: corticosterone (stress hormone), Eplerenone: mineralocorticoid receptor (MR) antagonist, Mifepristone: glucocorticoid receptor (GR) antagonist >
To understand the changes in astrocyte functions caused by childhood stress, the research team used mice models with early social deprivation, and discovered that stress hormones bind to the glucocorticoid receptors (GRs) of astrocytes. This significantly increased the expression of Mer tyrosine kinase (MERK), which plays an essential role in astrocyte phagocytosis. Surprisingly, out of the various neurons in the cerebral cortex, astrocytes would eliminate only the excitatory synapses of specific neurons. The team found that this builds abnormal neural networks, which can lead to complex behavioral abnormalities such as social deficiencies and depression in adulthood.
The team also observed that microglia, which also play an important role in cerebral immunity, did not contribute to synapse removal in the mice models with early social deprivation. This confirms that the response to stress hormones during childhood is specifically astrocyte-mediated.
To find out whether these results are also applicable in humans, the research team used a brain organoid grown from human-induced pluripotent stem cells to observe human responses to stress hormones. The team observed that the stress hormones induced astrocyte GRs and phagocyte activation in the human brain organoid as well, and confirmed that the astrocytes subsequently eliminated excessive amounts of excitatory synapses. By showing that mice and humans both showed the same synapse control mechanism in response to stress, the team suggested that this discovery is applicable to mental disorders in humans.
< Figure 2. A schematic diagram of the study published in Immunity. Excessive stress hormone secretion in childhood increases the expression of the MERTK phagocytic receptor through the glucocorticoid receptor (GR) of astrocytes, resulting in excessive elimination of excitatory synapses. Excessive synaptic elimination by astrocytes during brain development causes permanent damage to brain circuits, resulting in abnormal neural activity in the adult brain and psychiatric behaviors such as depression and anti-social tendencies. >
Prof. Won-Suk Chung said, “Until now, we did not know the exact mechanism for how childhood stress caused brain diseases. This research was the first to show that the excessive phagocytosis of astrocytes could be an important cause of such diseases.” He added, “In the future, controlling the immune response of astrocytes will be used as a fundamental target for understanding and treating brain diseases.”
This research, written by co-first authors Youkyeong Byun (Ph.D. candidate) and Nam-Shik Kim (post-doctoral associate) from the KAIST Department of Biological Sciences, was published in the internationally renowned journal Immunity, a sister magazine of Cell and one of the best journal in the field of immunology, on July 31 under the title "Stress induces behavioral abnormalities by increasing expression of phagocytic receptor MERTK in astrocytes to promote synapse phagocytosis."
This work was supported by a National Research Foundation of Korea grant, the Korea Health Industry Development Institute (KHIDI), and the Korea Dementia Research Center (KDRC).
2023.08.04 View 8169 -
 A New Therapeutic Drug for Alzheimer’s Disease without Inflammatory Side Effects
Although Aduhelm, a monoclonal antibody targeting amyloid beta (Aβ), recently became the first US FDA approved drug for Alzheimer’s disease (AD) based on its ability to decrease Aβ plaque burden in AD patients, its effect on cognitive improvement is still controversial. Moreover, about 40% of the patients treated with this antibody experienced serious side effects including cerebral edemas (ARIA-E) and hemorrhages (ARIA-H) that are likely related to inflammatory responses in the brain when the Aβ antibody binds Fc receptors (FCR) of immune cells such as microglia and macrophages.
These inflammatory side effects can cause neuronal cell death and synapse elimination by activated microglia, and even have the potential to exacerbate cognitive impairment in AD patients. Thus, current Aβ antibody-based immunotherapy holds the inherent risk of doing more harm than good due to their inflammatory side effects.
To overcome these problems, a team of researchers at KAIST in South Korea has developed a novel fusion protein drug, αAβ-Gas6, which efficiently eliminates Aβ via an entirely different mechanism than Aβ antibody-based immunotherapy. In a mouse model of AD, αAβ-Gas6 not only removed Aβ with higher potency, but also circumvented the neurotoxic inflammatory side effects associated with conventional antibody treatments.
Their findings were published on August 4 in Nature Medicine.
Schematic of a chimeric Gas6 fusion protein. A single chain variable fragment (scFv) of
an Amyloid β (Aβ)-targeting monoclonal antibody is fused with a truncated receptor binding
domain of Gas6, a bridging molecule for the clearance of dead cells via TAM (TYRO3, AXL,
and MERTK) receptors, which are expressed by microglia and astrocytes.
“FcR activation by Aβ targeting antibodies induces microglia-mediated Aβ phagocytosis, but it also produces inflammatory signals, inevitably damaging brain tissues,” said paper authors Chan Hyuk Kim and Won-Suk Chung, associate professors in the Department of Biological Sciences at KAIST.
“Therefore, we utilized efferocytosis, a cellular process by which dead cells are removed by phagocytes as an alternative pathway for the clearance of Aβ in the brain,” Prof. Kim and Chung said. “Efferocytosis is accompanied by anti-inflammatory responses to maintain tissue homeostasis. To exploit this process, we engineered Gas6, a soluble adaptor protein that mediates efferocytosis via TAM phagocytic receptors in such a way that its target specificity was redirected from dead cells to Aβ plaques.”
The professors and their team demonstrated that the resulting αAβ-Gas6 induced Aβ engulfment by activating not only microglial but also astrocytic phagocytosis since TAM phagocytic receptors are highly expressed by these two major phagocytes in the brain. Importantly, αAβ-Gas6 promoted the robust uptake of Aβ without showing any signs of inflammation and neurotoxicity, which contrasts sharply with the treatment using an Aβ monoclonal antibody. Moreover, they showed that αAβ-Gas6 substantially reduced excessive synapse elimination by microglia, consequently leading to better behavioral rescues in AD model mice.
“By using a mouse model of cerebral amyloid angiopathy (CAA), a cerebrovascular disorder caused by the deposition of Aβ within the walls of the brain’s blood vessels, we also showed that the intrathecal administration of Gas6 fusion protein significantly eliminated cerebrovascular amyloids, along with a reduction of microhemorrhages. These data demonstrate that aAb-Gas6 is a potent therapeutic agent in eliminating Aβ without exacerbating CAA-related microhemorrhages.”
The resulting αAβ-Gas6 clears Aβ oligomers and fibrils without causing neurotoxicity (a-b, neurons: red, and fragmented axons: yellow) and proinflammatory responses (c, TNF release), which are conversely exacerbated by the treatment of an Aβ-targeting monoclonal antibody (Aducanumab).
Professors Kim and Chung noted, “We believe our approach can be a breakthrough in treating AD without causing inflammatory side effects and synapse loss. Our approach holds promise as a novel therapeutic platform that is applicable to more than AD. By modifying the target-specificity of the fusion protein, the Gas6-fusion protein can be applied to various neurological disorders as well as autoimmune diseases affected by toxic molecules that should be removed without causing inflammatory responses.”
The number and total area of Aβ plaques (Thioflavin-T, green) were significantly reduced in αAβ-Gas6-treated AD mouse brains compared to Aducanumab-treated ones (a, b). The cognitive functions of AD model mice were significantly rescued by αAβ-Gas6 treatment, whereas Aducanumab-treated AD mice showed partial rescue in these cognitive tests (c-e).
Professors Kim and Chung founded “Illimis Therapeutics” based on this strategy of designing chimeric Gas6 fusion proteins that would remove toxic aggregates from the nervous system. Through this company, they are planning to further develop various Gas6-fusion proteins not only for Ab but also for Tau to treat AD symptoms.
This work was supported by KAIST and the Korea Health Technology R&D Project that was administered by the Korea Health Industry Development Institute (KHIDI) and the Korea Dementia Research Center (KDRC) funded by the Ministry of Health & Welfare (MOHW) and the Ministry of Science and ICT (MSIT), and KAIST.
Other contributors include Hyuncheol Jung and Se Young Lee, Sungjoon Lim, Hyeong Ryeol Choi, Yeseong Choi, Minjin Kim, Segi Kim, the Department of Biological Sciences, and the Korea Advanced Institute of Science and Technology (KAIST).
To receive more up-to-date information on this new development, follow “Illimis Therapeutics” on twitter @Illimistx.
2022.08.05 View 13419
A New Therapeutic Drug for Alzheimer’s Disease without Inflammatory Side Effects
Although Aduhelm, a monoclonal antibody targeting amyloid beta (Aβ), recently became the first US FDA approved drug for Alzheimer’s disease (AD) based on its ability to decrease Aβ plaque burden in AD patients, its effect on cognitive improvement is still controversial. Moreover, about 40% of the patients treated with this antibody experienced serious side effects including cerebral edemas (ARIA-E) and hemorrhages (ARIA-H) that are likely related to inflammatory responses in the brain when the Aβ antibody binds Fc receptors (FCR) of immune cells such as microglia and macrophages.
These inflammatory side effects can cause neuronal cell death and synapse elimination by activated microglia, and even have the potential to exacerbate cognitive impairment in AD patients. Thus, current Aβ antibody-based immunotherapy holds the inherent risk of doing more harm than good due to their inflammatory side effects.
To overcome these problems, a team of researchers at KAIST in South Korea has developed a novel fusion protein drug, αAβ-Gas6, which efficiently eliminates Aβ via an entirely different mechanism than Aβ antibody-based immunotherapy. In a mouse model of AD, αAβ-Gas6 not only removed Aβ with higher potency, but also circumvented the neurotoxic inflammatory side effects associated with conventional antibody treatments.
Their findings were published on August 4 in Nature Medicine.
Schematic of a chimeric Gas6 fusion protein. A single chain variable fragment (scFv) of
an Amyloid β (Aβ)-targeting monoclonal antibody is fused with a truncated receptor binding
domain of Gas6, a bridging molecule for the clearance of dead cells via TAM (TYRO3, AXL,
and MERTK) receptors, which are expressed by microglia and astrocytes.
“FcR activation by Aβ targeting antibodies induces microglia-mediated Aβ phagocytosis, but it also produces inflammatory signals, inevitably damaging brain tissues,” said paper authors Chan Hyuk Kim and Won-Suk Chung, associate professors in the Department of Biological Sciences at KAIST.
“Therefore, we utilized efferocytosis, a cellular process by which dead cells are removed by phagocytes as an alternative pathway for the clearance of Aβ in the brain,” Prof. Kim and Chung said. “Efferocytosis is accompanied by anti-inflammatory responses to maintain tissue homeostasis. To exploit this process, we engineered Gas6, a soluble adaptor protein that mediates efferocytosis via TAM phagocytic receptors in such a way that its target specificity was redirected from dead cells to Aβ plaques.”
The professors and their team demonstrated that the resulting αAβ-Gas6 induced Aβ engulfment by activating not only microglial but also astrocytic phagocytosis since TAM phagocytic receptors are highly expressed by these two major phagocytes in the brain. Importantly, αAβ-Gas6 promoted the robust uptake of Aβ without showing any signs of inflammation and neurotoxicity, which contrasts sharply with the treatment using an Aβ monoclonal antibody. Moreover, they showed that αAβ-Gas6 substantially reduced excessive synapse elimination by microglia, consequently leading to better behavioral rescues in AD model mice.
“By using a mouse model of cerebral amyloid angiopathy (CAA), a cerebrovascular disorder caused by the deposition of Aβ within the walls of the brain’s blood vessels, we also showed that the intrathecal administration of Gas6 fusion protein significantly eliminated cerebrovascular amyloids, along with a reduction of microhemorrhages. These data demonstrate that aAb-Gas6 is a potent therapeutic agent in eliminating Aβ without exacerbating CAA-related microhemorrhages.”
The resulting αAβ-Gas6 clears Aβ oligomers and fibrils without causing neurotoxicity (a-b, neurons: red, and fragmented axons: yellow) and proinflammatory responses (c, TNF release), which are conversely exacerbated by the treatment of an Aβ-targeting monoclonal antibody (Aducanumab).
Professors Kim and Chung noted, “We believe our approach can be a breakthrough in treating AD without causing inflammatory side effects and synapse loss. Our approach holds promise as a novel therapeutic platform that is applicable to more than AD. By modifying the target-specificity of the fusion protein, the Gas6-fusion protein can be applied to various neurological disorders as well as autoimmune diseases affected by toxic molecules that should be removed without causing inflammatory responses.”
The number and total area of Aβ plaques (Thioflavin-T, green) were significantly reduced in αAβ-Gas6-treated AD mouse brains compared to Aducanumab-treated ones (a, b). The cognitive functions of AD model mice were significantly rescued by αAβ-Gas6 treatment, whereas Aducanumab-treated AD mice showed partial rescue in these cognitive tests (c-e).
Professors Kim and Chung founded “Illimis Therapeutics” based on this strategy of designing chimeric Gas6 fusion proteins that would remove toxic aggregates from the nervous system. Through this company, they are planning to further develop various Gas6-fusion proteins not only for Ab but also for Tau to treat AD symptoms.
This work was supported by KAIST and the Korea Health Technology R&D Project that was administered by the Korea Health Industry Development Institute (KHIDI) and the Korea Dementia Research Center (KDRC) funded by the Ministry of Health & Welfare (MOHW) and the Ministry of Science and ICT (MSIT), and KAIST.
Other contributors include Hyuncheol Jung and Se Young Lee, Sungjoon Lim, Hyeong Ryeol Choi, Yeseong Choi, Minjin Kim, Segi Kim, the Department of Biological Sciences, and the Korea Advanced Institute of Science and Technology (KAIST).
To receive more up-to-date information on this new development, follow “Illimis Therapeutics” on twitter @Illimistx.
2022.08.05 View 13419 -
 Astrocytes Eat Connections to Maintain Plasticity in Adult Brains
Developing brains constantly sprout new neuronal connections called synapses as they learn and remember. Important connections — the ones that are repeatedly introduced, such as how to avoid danger — are nurtured and reinforced, while connections deemed unnecessary are pruned away. Adult brains undergo similar pruning, but it was unclear how or why synapses in the adult brain get eliminated.
Now, a team of KAIST researchers has found the mechanism underlying plasticity and, potentially, neurological disorders in adult brains. They published their findings on December 23 in Nature.
“Our findings have profound implications for our understanding of how neural circuits change during learning and memory, as well as in diseases,” said paper author Won-Suk Chung, an assistant professor in the Department of Biological Sciences at KAIST. “Changes in synapse number have strong association with the prevalence of various neurological disorders, such as autism spectrum disorder, schizophrenia, frontotemporal dementia, and several forms of seizures.”
Gray matter in the brain contains microglia and astrocytes, two complementary cells that, among other things, support neurons and synapses. Microglial are a frontline immunity defense, responsible for eating pathogens and dead cells, and astrocytes are star-shaped cells that help structure the brain and maintain homeostasis by helping to control signaling between neurons. According to Professor Chung, it is generally thought that microglial eat synapses as part of its clean-up effort in a process known as phagocytosis.
“Using novel tools, we show that, for the first time, it is astrocytes and not microglia that constantly eliminate excessive and unnecessary adult excitatory synaptic connections in response to neuronal activity,” Professor Chung said. “Our paper challenges the general consensus in this field that microglia are the primary synapse phagocytes that control synapse numbers in the brain.”
Professor Chung and his team developed a molecular sensor to detect synapse elimination by glial cells and quantified how often and by which type of cell synapses were eliminated. They also deployed it in a mouse model without MEGF10, the gene that allows astrocytes to eliminate synapses. Adult animals with this defective astrocytic phagocytosis had unusually increased excitatory synapse numbers in the hippocampus. Through a collaboration with Dr. Hyungju Park at KBRI, they showed that these increased excitatory synapses are functionally impaired, which cause defective learning and memory formation in MEGF10 deleted animals.
“Through this process, we show that, at least in the adult hippocampal CA1 region, astrocytes are the major player in eliminating synapses, and this astrocytic function is essential for controlling synapse number and plasticity,” Chung said.
Professor Chung noted that researchers are only beginning to understand how synapse elimination affects maturation and homeostasis in the brain. In his group’s preliminary data in other brain regions, it appears that each region has different rates of synaptic elimination by astrocytes. They suspect a variety of internal and external factors are influencing how astrocytes modulate each regional circuit, and plan to elucidate these variables.
“Our long-term goal is understanding how astrocyte-mediated synapse turnover affects the initiation and progression of various neurological disorders,” Professor Chung said. “It is intriguing to postulate that modulating astrocytic phagocytosis to restore synaptic connectivity may be a novel strategy in treating various brain disorders.”
This work was supported by the Samsung Science & Technology Foundation, the National Research Foundation of Korea, and the Korea Brain Research Institute basic research program.
Other contributors include Joon-Hyuk Lee and Se Young Lee, Department of Biological Sciences, Korea Advanced Institute of Science and Technology (KAIST); Ji-young Kim, Hyoeun Lee and Hyungju Park; Research Group for Neurovascular Unit, Korea Brain Research Institute (KBRI); Seulgi Noh, and Ji Young Mun, Research Group for Neural Circuit, KBRI. Kim, Noh and Park are also affiliated with the Department of Brain and Cognitive Sciences, Daegu Gyeongbuk Institute of Science and Technology (DGIST).
-Profile
Professor Won-Suk Chung
Department of Biological Sciences
Gliabiology Lab (https://www.kaistglia.org/)
KAIST
-Publication
"Astrocytes phagocytose adult hippocampal synapses for circuit homeostasis"
https://doi.org/10.1038/s41586-020-03060-3
2020.12.24 View 12815
Astrocytes Eat Connections to Maintain Plasticity in Adult Brains
Developing brains constantly sprout new neuronal connections called synapses as they learn and remember. Important connections — the ones that are repeatedly introduced, such as how to avoid danger — are nurtured and reinforced, while connections deemed unnecessary are pruned away. Adult brains undergo similar pruning, but it was unclear how or why synapses in the adult brain get eliminated.
Now, a team of KAIST researchers has found the mechanism underlying plasticity and, potentially, neurological disorders in adult brains. They published their findings on December 23 in Nature.
“Our findings have profound implications for our understanding of how neural circuits change during learning and memory, as well as in diseases,” said paper author Won-Suk Chung, an assistant professor in the Department of Biological Sciences at KAIST. “Changes in synapse number have strong association with the prevalence of various neurological disorders, such as autism spectrum disorder, schizophrenia, frontotemporal dementia, and several forms of seizures.”
Gray matter in the brain contains microglia and astrocytes, two complementary cells that, among other things, support neurons and synapses. Microglial are a frontline immunity defense, responsible for eating pathogens and dead cells, and astrocytes are star-shaped cells that help structure the brain and maintain homeostasis by helping to control signaling between neurons. According to Professor Chung, it is generally thought that microglial eat synapses as part of its clean-up effort in a process known as phagocytosis.
“Using novel tools, we show that, for the first time, it is astrocytes and not microglia that constantly eliminate excessive and unnecessary adult excitatory synaptic connections in response to neuronal activity,” Professor Chung said. “Our paper challenges the general consensus in this field that microglia are the primary synapse phagocytes that control synapse numbers in the brain.”
Professor Chung and his team developed a molecular sensor to detect synapse elimination by glial cells and quantified how often and by which type of cell synapses were eliminated. They also deployed it in a mouse model without MEGF10, the gene that allows astrocytes to eliminate synapses. Adult animals with this defective astrocytic phagocytosis had unusually increased excitatory synapse numbers in the hippocampus. Through a collaboration with Dr. Hyungju Park at KBRI, they showed that these increased excitatory synapses are functionally impaired, which cause defective learning and memory formation in MEGF10 deleted animals.
“Through this process, we show that, at least in the adult hippocampal CA1 region, astrocytes are the major player in eliminating synapses, and this astrocytic function is essential for controlling synapse number and plasticity,” Chung said.
Professor Chung noted that researchers are only beginning to understand how synapse elimination affects maturation and homeostasis in the brain. In his group’s preliminary data in other brain regions, it appears that each region has different rates of synaptic elimination by astrocytes. They suspect a variety of internal and external factors are influencing how astrocytes modulate each regional circuit, and plan to elucidate these variables.
“Our long-term goal is understanding how astrocyte-mediated synapse turnover affects the initiation and progression of various neurological disorders,” Professor Chung said. “It is intriguing to postulate that modulating astrocytic phagocytosis to restore synaptic connectivity may be a novel strategy in treating various brain disorders.”
This work was supported by the Samsung Science & Technology Foundation, the National Research Foundation of Korea, and the Korea Brain Research Institute basic research program.
Other contributors include Joon-Hyuk Lee and Se Young Lee, Department of Biological Sciences, Korea Advanced Institute of Science and Technology (KAIST); Ji-young Kim, Hyoeun Lee and Hyungju Park; Research Group for Neurovascular Unit, Korea Brain Research Institute (KBRI); Seulgi Noh, and Ji Young Mun, Research Group for Neural Circuit, KBRI. Kim, Noh and Park are also affiliated with the Department of Brain and Cognitive Sciences, Daegu Gyeongbuk Institute of Science and Technology (DGIST).
-Profile
Professor Won-Suk Chung
Department of Biological Sciences
Gliabiology Lab (https://www.kaistglia.org/)
KAIST
-Publication
"Astrocytes phagocytose adult hippocampal synapses for circuit homeostasis"
https://doi.org/10.1038/s41586-020-03060-3
2020.12.24 View 12815