본문 바로가기
대메뉴 바로가기
KAIST
Newsletter Vol.28
Receive KAIST news by email!
View
Subscribe
Close
Type your e-mail address here.
Subscribe
Close
KAIST
NEWS
유틸열기
홈페이지 통합검색
-
검색
KOREAN
메뉴 열기
quantum+properties
by recently order
by view order
KAIST Proposes AI Training Method that will Drastically Shorten Time for Complex Quantum Mechanical Calculations
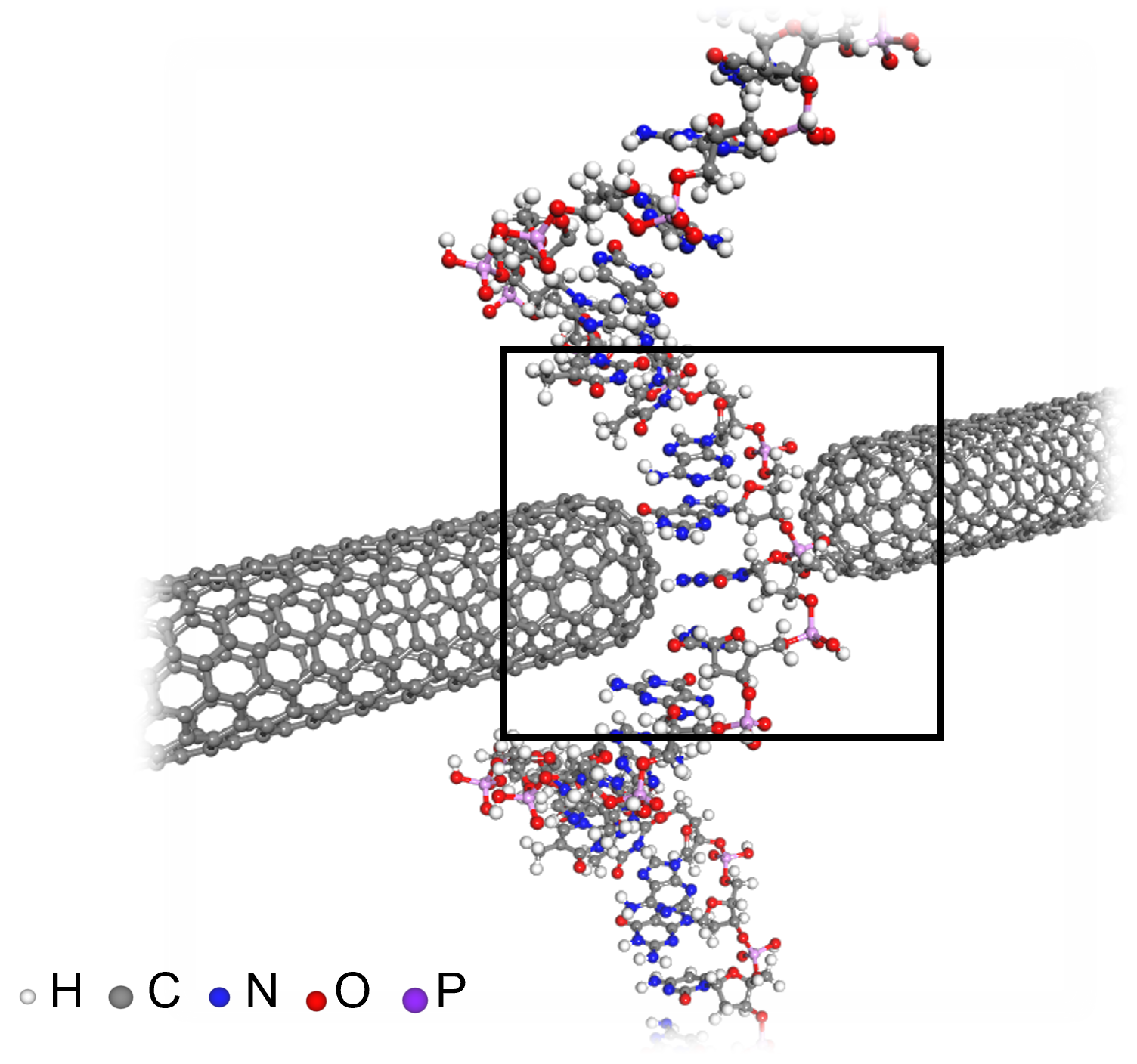
- Professor Yong-Hoon Kim's team from the School of Electrical Engineering succeeded for the first time in accelerating quantum mechanical electronic structure calculations using a convolutional neural network (CNN) model - Presenting an AI learning principle of quantum mechanical 3D chemical bonding information, the work is expected to accelerate the computer-assisted designing of next-generation materials and devices The close relationship between AI and high-performance scientific computing can be seen in the fact that both the 2024 Nobel Prizes in Physics and Chemistry were awarded to scientists for their AI-related research contributions in their respective fields of study. KAIST researchers succeeded in dramatically reducing the computation time for highly sophisticated quantum mechanical computer simulations by predicting atomic-level chemical bonding information distributed in 3D space using a novel AI approach. KAIST (President Kwang-Hyung Lee) announced on the 30th of October that Professor Yong-Hoon Kim's team from the School of Electrical Engineering developed a 3D computer vision artificial neural network-based computation methodology that bypasses the complex algorithms required for atomic-level quantum mechanical calculations traditionally performed using supercomputers to derive the properties of materials. < Figure 1. Various methodologies are utilized in the simulation of materials and materials, such as quantum mechanical calculations at the nanometer (nm) level, classical mechanical force fields at the scale of tens to hundreds of nanometers, continuum dynamics calculations at the macroscopic scale, and calculations that mix simulations at different scales. These simulations are already playing a key role in a wide range of basic research and application development fields in combination with informatics techniques. Recently, there have been active efforts to introduce machine learning techniques to radically accelerate simulations, but research on introducing machine learning techniques to quantum mechanical electronic structure calculations, which form the basis of high-scale simulations, is still insufficient. > The quantum mechanical density functional theory (DFT) calculations using supercomputers have become an essential and standard tool in a wide range of research and development fields, including advanced materials and drug design, as they allow fast and accurate prediction of material properties. *Density functional theory (DFT): A representative theory of ab initio (first principles) calculations that calculate quantum mechanical properties from the atomic level. However, practical DFT calculations require generating 3D electron density and solving quantum mechanical equations through a complex, iterative self-consistent field (SCF)* process that must be repeated tens to hundreds of times. This restricts its application to systems with only a few hundred to a few thousand atoms. *Self-consistent field (SCF): A scientific computing method widely used to solve complex many-body problems that must be described by a number of interconnected simultaneous differential equations. Professor Yong-Hoon Kim’s research team questioned whether recent advancements in AI techniques could be used to bypass the SCF process. As a result, they developed the DeepSCF model, which accelerates calculations by learning chemical bonding information distributed in a 3D space using neural network algorithms from the field of computer vision. < Figure 2. The deepSCF methodology developed in this study provides a way to rapidly accelerate DFT calculations by avoiding the self-consistent field process (orange box) that had to be performed repeatedly in traditional quantum mechanical electronic structure calculations through artificial neural network techniques (green box). The self-consistent field process is a process of predicting the 3D electron density, constructing the corresponding potential, and then solving the quantum mechanical Cohn-Sham equations, repeating tens to hundreds of times. The core idea of the deepSCF methodology is that the residual electron density (δρ), which is the difference between the electron density (ρ) and the sum of the electron densities of the constituent atoms (ρ0), corresponds to chemical bonding information, so the self-consistent field process is replaced with a 3D convolutional neural network model. > The research team focused on the fact that, according to density functional theory, electron density contains all quantum mechanical information of electrons, and that the residual electron density — the difference between the total electron density and the sum of the electron densities of the constituent atoms — contains chemical bonding information. They used this as the target for machine learning. They then adopted a dataset of organic molecules with various chemical bonding characteristics, and applied random rotations and deformations to the atomic structures of these molecules to further enhance the model’s accuracy and generalization capabilities. Ultimately, the research team demonstrated the validity and efficiency of the DeepSCF methodology on large, complex systems. < Figure 3. An example of applying the deepSCF methodology to a carbon nanotube-based DNA sequence analysis device model (top left). In addition to classical mechanical interatomic forces (bottom right), the residual electron density (top right) and quantum mechanical electronic structure properties such as the electronic density of states (DOS) (bottom left) containing information on chemical bonding are rapidly predicted with an accuracy corresponding to the standard DFT calculation results that perform the SCF process. > Professor Yong-Hoon Kim, who supervised the research, explained that his team had found a way to map quantum mechanical chemical bonding information in a 3D space onto artificial neural networks. He noted, “Since quantum mechanical electron structure calculations underpin materials simulations across all scales, this research establishes a foundational principle for accelerating material calculations using artificial intelligence.” Ryong-Gyu Lee, a PhD candidate in the School of Electrical Engineering, served as the first author of this research, which was published online on October 24 in Npj Computational Materials, a prestigious journal in the field of material computation. (Paper title: “Convolutional network learning of self-consistent electron density via grid-projected atomic fingerprints”) This research was conducted with support from the KAIST High-Risk Research Program for Graduate Students and the National Research Foundation of Korea’s Mid-career Researcher Support Program.
2024.10.30
View 7043
<<
첫번째페이지
<
이전 페이지
1
>
다음 페이지
>>
마지막 페이지 1