research
Cancer is characterized by abnormal metabolic processes different from those of normal cells. Therefore, cancer metabolism has been extensively studied to develop effective diagnosis and treatment strategies. Notable achievements of cancer metabolism studies include the discovery of oncometabolites* and the approval of anticancer drugs by the U.S. Food and Drug Administration (FDA) that target enzymes associated with oncometabolites. Approved anticancer drugs such as ‘Tibsovo (active ingredient: ivosidenib)’ and ‘Idhifa (active ingredient: enasidenib)’ are both used for the treatment of acute myeloid leukemia. Despite such achievements, studying cancer metabolism, especially oncometabolites, remains challenging due to time-consuming and expensive methodologies such as metabolomics. Thus, the number of confirmed oncometabolites is very small although a relatively large number of cancer-associated gene mutations have been well studied.
*Oncometabolite: A metabolite that shows pro-oncogenic function when abnormally accumulated in cancer cells. An oncometabolite is often generated as a result of gene mutations, and this accumulation promotes the growth and survival of cancer cells. Representative oncometabolites include 2-hydroxyglutarate, succinate, and fumarate.
On March 18th, a KAIST research team led by Professor Hyun Uk Kim from the Department of Chemical and Biomolecular Engineering developed a computational workflow that systematically predicts metabolites and metabolic pathways associated with somatic mutations in cancer through collaboration with research teams under Prof Youngil Koh, Prof. Hongseok Yun, and Prof. Chang Wook Jeong from Seoul National University Hospital.
The research teams have successfully reconstructed patient-specific genome-scale metabolic models (GEMs)* for 1,043 cancer patients across 24 cancer types by integrating publicly available cancer patients’ transcriptome data (i.e., from international cancer genome consortiums such as PCAWG and TCGA) into a generic human GEM. The resulting patient-specific GEMs make it possible to predict each patient’s metabolic phenotypes.
*Genome-scale metabolic model (GEM): A computational model that mathematically describes all of the biochemical reactions that take place inside a cell. It allows for the prediction of the cell’s metabolic phenotypes under various genetic and/or environmental conditions.
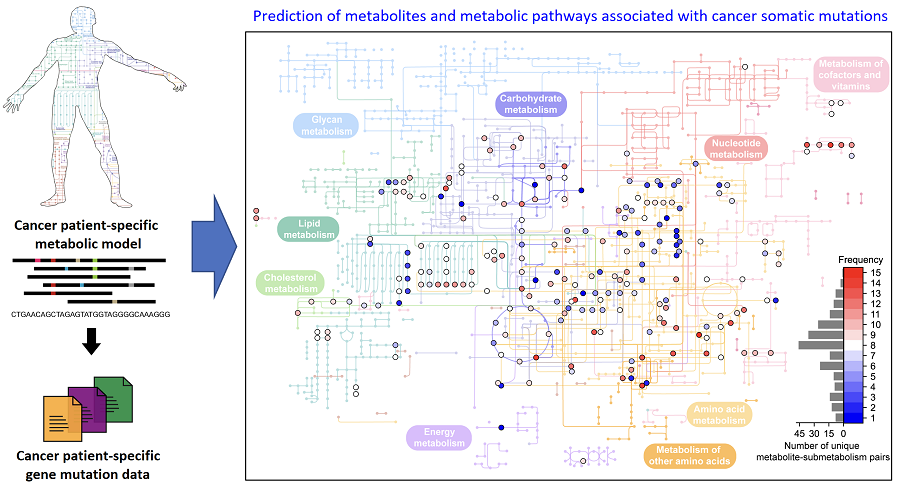
< Figure 1. Schematic diagram of a computational methodology for predicting metabolites and metabolic pathways associated with cancer somatic mutations. of a computational methodology for predicting metabolites and metabolic pathways associated with cancer somatic mutations. >
The team developed a four-step computational workflow using the patient-specific GEMs from 1,043 cancer patients and somatic mutation data obtained from the corresponding cancer patients. This workflow begins with the calculation of the flux-sum value of each metabolite by simulating the patient-specific GEMs. The flux-sum value quantifies the intracellular importance of a metabolite. Next, the workflow identifies metabolites that appear to be significantly associated with specific gene mutations through a statistical analysis of the predicted flux-sum data and the mutation data. Finally, the workflow selects altered metabolic pathways that significantly contribute to the biosynthesis of the predicted oncometabolite candidates, ultimately generating metabolite-gene-pathway sets as an output.
The two co-first authors, Dr. GaRyoung Lee (currently a postdoctoral fellow at the Dana-Farber Cancer Institute and Harvard Medical School) and Dr. Sang Mi Lee (currently a postdoctoral fellow at Harvard Medical School) said, “The computational workflow developed can systematically predict how genetic mutations affect cellular metabolism through metabolic pathways. Importantly, it can easily be applied to different types of cancer based on the mutation and transcriptome data of cancer patient cohorts.”
Prof. Kim said, “The computational workflow and its resulting prediction outcomes will serve as the groundwork for identifying novel oncometabolites and for facilitating the development of various treatment and diagnosis strategies”.
This study, which was supported by the National Research Foundation of Korea, has been published online in Genome Biology, a representative journal in the field of biotechnology and genetics, under the title "Prediction of metabolites associated with somatic mutations in cancers by using genome‑scale metabolic models and mutation data".
-
research KAIST Develops Virtual Staining Technology for 3D Histopathology
Moving beyond traditional methods of observing thinly sliced and stained cancer tissues, a collaborative international research team led by KAIST has successfully developed a groundbreaking technology. This innovation uses advanced optical techniques combined with an artificial intelligence-based deep learning algorithm to create realistic, virtually stained 3D images of cancer tissue without the need for serial sectioning nor staining. This breakthrough is anticipated to pave the way for next-g
2025-05-26 -
research KAIST provides a comprehensive resource on microbial cell factories for sustainable chemical production
In silico analysis of five industrial microorganisms identifies optimal strains and metabolic engineering strategies for producing 235 valuable chemicals Climate change and the depletion of fossil fuels have raised the global need for sustainable chemical production. In response to these environmental challenges, microbial cell factories are gaining attention as eco-friendly platforms for producing chemicals using renewable resources, while metabolic engineering technologies to enhance these
2025-03-27 -
research KAIST Discovers Molecular Switch that Reverses Cancerous Transformation at the Critical Moment of Transition
< (From left) PhD student Seoyoon D. Jeong, (bottom) Professor Kwang-Hyun Cho, (top) Dr. Dongkwan Shin, Dr. Jeong-Ryeol Gong > Professor Kwang-Hyun Cho’s research team has recently been highlighted for their work on developing an original technology for cancer reversal treatment that does not kill cancer cells but only changes their characteristics to reverse them to a state similar to normal cells. This time, they have succeeded in revealing for the first time that a molecular
2025-02-05 -
research KAIST Develops Foundational Technology to Revert Cancer Cells to Normal Cells
Despite the development of numerous cancer treatment technologies, the common goal of current cancer therapies is to eliminate cancer cells. This approach, however, faces fundamental limitations, including cancer cells developing resistance and returning, as well as severe side effects from the destruction of healthy cells. < (From top left) Bio and Brain Engineering PhD candidates Juhee Kim, Jeong-Ryeol Gong, Chun-Kyung Lee, and Hoon-Min Kim posed for a group photo with Professor Kwang-
2024-12-23 -
research Genome Sequencing Unveils Mutational Impacts of Radiation on Mammalian Cells
Recent release of the waste water from Japan's Fukushima nuclear disaster stirred apprehension regarding the health implications of radiation exposure. Classified as a Group 1 carcinogen, ionizing radiation has long been associated with various cancers and genetic disorders, as evidenced by survivors and descendants of atomic bombings and the Chernobyl disaster. Despite much smaller amount, we remain consistently exposed to low levels of radiation in everyday life and medical procedures. Radi
2024-02-15