prediction
-
 KAIST Accelerates Synthetic Microbe Design by Discovering Novel Enzymes Using AI
< (From left) Professor Sang Yup Lee of the Department of Chemical and Biomolecular Engineering (top), Hongkeun Ji, PhD candidate of the Department of Chemical and Biomolecular Engineering (top), Ha Rim Kim, PhD candidate of the Department of Chemical and Biomolecular Engineering, and Dr. Gi Bae Kim of the BioProcess Engineering Research Center >
Enzymes are proteins that catalyze biochemical reactions within cells and play a pivotal role in metabolic processes. Accordingly, identifying the functions of novel enzymes is a critical task in the construction of microbial cell factories.
A KAIST research team has leveraged artificial intelligence (AI) to design novel enzymes that do not exist in nature, significantly accelerating microbial cell factory development and boosting the potential for next-generation biotechnological applications such as drug development and biofuel production.
KAIST (represented by President Kwang-Hyung Lee) announced on the 21st of April that Distinguished Professor Sang Yup Lee and his team from the Department of Chemical and Biomolecular Engineering have published a review titled “Enzyme Functional Classification Using Artificial Intelligence,” which outlines the advancement of AI-based enzyme function prediction technologies and analyzes how AI has contributed to the discovery and design of new enzymes.
Professor Lee’s team systematically reviewed the development of enzyme function prediction technologies utilizing machine learning and deep learning, offering a comprehensive analysis.
From sequence similarity-based prediction methods to the integration of convolutional neural networks (CNNs), recurrent neural networks (RNNs), graph neural networks (GNNs), and transformer-based large language models, the paper covers a broad range of AI applications. It analyzes how these technologies extract meaningful information from protein sequences and enhance prediction accuracy.
In particular, enzyme function prediction using deep learning goes beyond simple sequence similarity analysis. By automatically extracting structural and evolutionary features embedded in amino acid sequences, deep learning enables more precise predictions of catalytic functions.
This highlights the unique advantages of AI models compared to traditional bioinformatics approaches.
Moreover, the review suggests that the advancement of generative AI will move future research beyond predicting existing functions to generating entirely new enzymes with functions not found in nature. This shift is expected to profoundly impact the trajectory of biotechnology and synthetic biology.
< Figure 1. Extraction of enzyme characteristics and function prediction using various deep learning structures >
Ha Rim Kim, a Ph.D. candidate and co-first author from the Department of Chemical and Biomolecular Engineering, stated, “AI-based enzyme function prediction and enzyme design are highly important across various fields including metabolic engineering, synthetic biology, and healthcare.”
Distinguished Professor Sang Yup Lee added, “AI-powered enzyme function prediction shows the potential to solve diverse biological problems and will significantly contribute to accelerating research across the entire field.”
The review was published on March 28 in Trends in Biotechnology, a leading biotechnology journal issued by Cell Press.
※ Title: Enzyme Functional Classification Using Artificial Intelligence
※DOI: https://doi.org/10.1016/j.tibtech.2025.03.003
※ Author Information: Ha Rim Kim (KAIST, Co-first author), Hongkeun Ji (KAIST, Co-first author), Gi Bae Kim (KAIST, Third author), Sang Yup Lee (KAIST, Corresponding author)
This research was supported by the Ministry of Science and ICT under the project Development of Core Technologies for Advanced Synthetic Biology to Lead the Bio-Manufacturing Industry (aimed at replacing petroleum-based chemicals), and also by joint support from the Ministry of Science and ICT and the Ministry of Health and Welfare for the project Development of Novel Antibiotic Structures Using Deep Learning-Based Synthetic Biology.
2025.04.07 View 3129
KAIST Accelerates Synthetic Microbe Design by Discovering Novel Enzymes Using AI
< (From left) Professor Sang Yup Lee of the Department of Chemical and Biomolecular Engineering (top), Hongkeun Ji, PhD candidate of the Department of Chemical and Biomolecular Engineering (top), Ha Rim Kim, PhD candidate of the Department of Chemical and Biomolecular Engineering, and Dr. Gi Bae Kim of the BioProcess Engineering Research Center >
Enzymes are proteins that catalyze biochemical reactions within cells and play a pivotal role in metabolic processes. Accordingly, identifying the functions of novel enzymes is a critical task in the construction of microbial cell factories.
A KAIST research team has leveraged artificial intelligence (AI) to design novel enzymes that do not exist in nature, significantly accelerating microbial cell factory development and boosting the potential for next-generation biotechnological applications such as drug development and biofuel production.
KAIST (represented by President Kwang-Hyung Lee) announced on the 21st of April that Distinguished Professor Sang Yup Lee and his team from the Department of Chemical and Biomolecular Engineering have published a review titled “Enzyme Functional Classification Using Artificial Intelligence,” which outlines the advancement of AI-based enzyme function prediction technologies and analyzes how AI has contributed to the discovery and design of new enzymes.
Professor Lee’s team systematically reviewed the development of enzyme function prediction technologies utilizing machine learning and deep learning, offering a comprehensive analysis.
From sequence similarity-based prediction methods to the integration of convolutional neural networks (CNNs), recurrent neural networks (RNNs), graph neural networks (GNNs), and transformer-based large language models, the paper covers a broad range of AI applications. It analyzes how these technologies extract meaningful information from protein sequences and enhance prediction accuracy.
In particular, enzyme function prediction using deep learning goes beyond simple sequence similarity analysis. By automatically extracting structural and evolutionary features embedded in amino acid sequences, deep learning enables more precise predictions of catalytic functions.
This highlights the unique advantages of AI models compared to traditional bioinformatics approaches.
Moreover, the review suggests that the advancement of generative AI will move future research beyond predicting existing functions to generating entirely new enzymes with functions not found in nature. This shift is expected to profoundly impact the trajectory of biotechnology and synthetic biology.
< Figure 1. Extraction of enzyme characteristics and function prediction using various deep learning structures >
Ha Rim Kim, a Ph.D. candidate and co-first author from the Department of Chemical and Biomolecular Engineering, stated, “AI-based enzyme function prediction and enzyme design are highly important across various fields including metabolic engineering, synthetic biology, and healthcare.”
Distinguished Professor Sang Yup Lee added, “AI-powered enzyme function prediction shows the potential to solve diverse biological problems and will significantly contribute to accelerating research across the entire field.”
The review was published on March 28 in Trends in Biotechnology, a leading biotechnology journal issued by Cell Press.
※ Title: Enzyme Functional Classification Using Artificial Intelligence
※DOI: https://doi.org/10.1016/j.tibtech.2025.03.003
※ Author Information: Ha Rim Kim (KAIST, Co-first author), Hongkeun Ji (KAIST, Co-first author), Gi Bae Kim (KAIST, Third author), Sang Yup Lee (KAIST, Corresponding author)
This research was supported by the Ministry of Science and ICT under the project Development of Core Technologies for Advanced Synthetic Biology to Lead the Bio-Manufacturing Industry (aimed at replacing petroleum-based chemicals), and also by joint support from the Ministry of Science and ICT and the Ministry of Health and Welfare for the project Development of Novel Antibiotic Structures Using Deep Learning-Based Synthetic Biology.
2025.04.07 View 3129 -
 KAIST Develops AI-Driven Performance Prediction Model to Advance Space Electric Propulsion Technology
< (From left) PhD candidate Youngho Kim, Professor Wonho Choe, and PhD candidate Jaehong Park from the Department of Nuclear and Quantum Engineering >
Hall thrusters, a key space technology for missions like SpaceX's Starlink constellation and NASA's Psyche asteroid mission, are high-efficiency electric propulsion devices using plasma technology*. The KAIST research team announced that the AI-designed Hall thruster developed for CubeSats will be installed on the KAIST-Hall Effect Rocket Orbiter (K-HERO) CubeSat to demonstrate its in-orbit performance during the fourth launch of the Korean Launch Vehicle called Nuri rocket (KSLV-2) scheduled for November this year.
*Plasma is one of the four states of matter, where gases are heated to high energies, causing them to separate into charged ions and electrons. Plasma is used not only in space electric propulsion but also in semiconductor manufacturing, display processes, and sterilization devices.
On February 3rd, the research team from the KAIST Department of Nuclear and Quantum Engineering’s Electric Propulsion Laboratory, led by Professor Wonho Choe, announced the development of an AI-based technique to accurately predict the performance of Hall thrusters, the engines of satellites and space probes.
Hall thrusters provide high fuel efficiency, requiring minimal propellant to achieve significant acceleration of spacecrafts or satellites while producing substantial thrust relative to power consumption. Due to these advantages, Hall thrusters are widely used in various space missions, including the formation flight of satellite constellations, deorbiting maneuvers for space debris mitigation, and deep space missions such as asteroid exploration.
As the space industry continues to grow during the NewSpace era, the demand for Hall thrusters suited to diverse missions is increasing. To rapidly develop highly efficient, mission-optimized Hall thrusters, it is essential to predict thruster performance accurately from the design phase.
However, conventional methods have limitations, as they struggle to handle the complex plasma phenomena within Hall thrusters or are only applicable under specific conditions, leading to lower prediction accuracy.
The research team developed an AI-based performance prediction technique with high accuracy, significantly reducing the time and cost associated with the iterative design, fabrication, and testing of thrusters. Since 2003, Professor Wonho Choe’s team has been leading research on electric propulsion development in Korea. The team applied a neural network ensemble model to predict thruster performance using 18,000 Hall thruster training data points generated from their in-house numerical simulation tool.
The in-house numerical simulation tool, developed to model plasma physics and thrust performance, played a crucial role in providing high-quality training data. The simulation’s accuracy was validated through comparisons with experimental data from ten KAIST in-house Hall thrusters, with an average prediction error of less than 10%.
< Figure 1. This research has been selected as the cover article for the March 2025 issue (Volume 7, Issue 3) of the AI interdisciplinary journal, Advanced Intelligent Systems. >
The trained neural network ensemble model acts as a digital twin, accurately predicting the Hall thruster performance within seconds based on thruster design variables.
Notably, it offers detailed analyses of performance parameters such as thrust and discharge current, accounting for Hall thruster design variables like propellant flow rate and magnetic field—factors that are challenging to evaluate using traditional scaling laws.
This AI model demonstrated an average prediction error of less than 5% for the in-house 700 W and 1 kW KAIST Hall thrusters and less than 9% for a 5 kW high-power Hall thruster developed by the University of Michigan and the U.S. Air Force Research Laboratory. This confirms the broad applicability of the AI prediction method across different power levels of Hall thrusters.
Professor Wonho Choe stated, “The AI-based prediction technique developed by our team is highly accurate and is already being utilized in the analysis of thrust performance and the development of highly efficient, low-power Hall thrusters for satellites and spacecraft. This AI approach can also be applied beyond Hall thrusters to various industries, including semiconductor manufacturing, surface processing, and coating, through ion beam sources.”
< Figure 2. The AI-based prediction technique developed by the research team accurately predicts thrust performance based on design variables, making it highly valuable for the development of high-efficiency Hall thrusters. The neural network ensemble processes design variables, such as channel geometry and magnetic field information, and outputs key performance metrics like thrust and prediction accuracy, enabling efficient thruster design and performance analysis. >
Additionally, Professor Choe mentioned, “The CubeSat Hall thruster, developed using the AI technique in collaboration with our lab startup—Cosmo Bee, an electric propulsion company—will be tested in orbit this November aboard the K-HERO 3U (30 x 10 x 10 cm) CubeSat, scheduled for launch on the fourth flight of the KSLV-2 Nuri rocket.”
This research was published online in Advanced Intelligent Systems on December 25, 2024 with PhD candidate Jaehong Park as the first author and was selected as the journal’s cover article, highlighting its innovation.
< Figure 3. Image of the 150 W low-power Hall thruster for small and micro satellites, developed in collaboration with Cosmo Bee and the KAIST team. The thruster will be tested in orbit on the K-HERO CubeSat during the KSLV-2 Nuri rocket’s fourth launch in Q4 2025. >
This research was supported by the National Research Foundation of Korea’s Space Pioneer Program (200mN High Thrust Electric Propulsion System Development).
(Paper Title: Predicting Performance of Hall Effect Ion Source Using Machine Learning, DOI: https://doi.org/10.1002/aisy.202400555 )
< Figure 4. Graphs of the predicted thrust and discharge current of KAIST’s 700 W Hall thruster using the AI model (HallNN). The left image shows the Hall thruster operating in KAIST Electric Propulsion Laboratory’s vacuum chamber, while the center and right graphs present the prediction results for thrust and discharge current based on anode mass flow rate. The red lines represent AI predictions, and the blue dots represent experimental results, with a prediction error of less than 5%. >
2025.02.03 View 5889
KAIST Develops AI-Driven Performance Prediction Model to Advance Space Electric Propulsion Technology
< (From left) PhD candidate Youngho Kim, Professor Wonho Choe, and PhD candidate Jaehong Park from the Department of Nuclear and Quantum Engineering >
Hall thrusters, a key space technology for missions like SpaceX's Starlink constellation and NASA's Psyche asteroid mission, are high-efficiency electric propulsion devices using plasma technology*. The KAIST research team announced that the AI-designed Hall thruster developed for CubeSats will be installed on the KAIST-Hall Effect Rocket Orbiter (K-HERO) CubeSat to demonstrate its in-orbit performance during the fourth launch of the Korean Launch Vehicle called Nuri rocket (KSLV-2) scheduled for November this year.
*Plasma is one of the four states of matter, where gases are heated to high energies, causing them to separate into charged ions and electrons. Plasma is used not only in space electric propulsion but also in semiconductor manufacturing, display processes, and sterilization devices.
On February 3rd, the research team from the KAIST Department of Nuclear and Quantum Engineering’s Electric Propulsion Laboratory, led by Professor Wonho Choe, announced the development of an AI-based technique to accurately predict the performance of Hall thrusters, the engines of satellites and space probes.
Hall thrusters provide high fuel efficiency, requiring minimal propellant to achieve significant acceleration of spacecrafts or satellites while producing substantial thrust relative to power consumption. Due to these advantages, Hall thrusters are widely used in various space missions, including the formation flight of satellite constellations, deorbiting maneuvers for space debris mitigation, and deep space missions such as asteroid exploration.
As the space industry continues to grow during the NewSpace era, the demand for Hall thrusters suited to diverse missions is increasing. To rapidly develop highly efficient, mission-optimized Hall thrusters, it is essential to predict thruster performance accurately from the design phase.
However, conventional methods have limitations, as they struggle to handle the complex plasma phenomena within Hall thrusters or are only applicable under specific conditions, leading to lower prediction accuracy.
The research team developed an AI-based performance prediction technique with high accuracy, significantly reducing the time and cost associated with the iterative design, fabrication, and testing of thrusters. Since 2003, Professor Wonho Choe’s team has been leading research on electric propulsion development in Korea. The team applied a neural network ensemble model to predict thruster performance using 18,000 Hall thruster training data points generated from their in-house numerical simulation tool.
The in-house numerical simulation tool, developed to model plasma physics and thrust performance, played a crucial role in providing high-quality training data. The simulation’s accuracy was validated through comparisons with experimental data from ten KAIST in-house Hall thrusters, with an average prediction error of less than 10%.
< Figure 1. This research has been selected as the cover article for the March 2025 issue (Volume 7, Issue 3) of the AI interdisciplinary journal, Advanced Intelligent Systems. >
The trained neural network ensemble model acts as a digital twin, accurately predicting the Hall thruster performance within seconds based on thruster design variables.
Notably, it offers detailed analyses of performance parameters such as thrust and discharge current, accounting for Hall thruster design variables like propellant flow rate and magnetic field—factors that are challenging to evaluate using traditional scaling laws.
This AI model demonstrated an average prediction error of less than 5% for the in-house 700 W and 1 kW KAIST Hall thrusters and less than 9% for a 5 kW high-power Hall thruster developed by the University of Michigan and the U.S. Air Force Research Laboratory. This confirms the broad applicability of the AI prediction method across different power levels of Hall thrusters.
Professor Wonho Choe stated, “The AI-based prediction technique developed by our team is highly accurate and is already being utilized in the analysis of thrust performance and the development of highly efficient, low-power Hall thrusters for satellites and spacecraft. This AI approach can also be applied beyond Hall thrusters to various industries, including semiconductor manufacturing, surface processing, and coating, through ion beam sources.”
< Figure 2. The AI-based prediction technique developed by the research team accurately predicts thrust performance based on design variables, making it highly valuable for the development of high-efficiency Hall thrusters. The neural network ensemble processes design variables, such as channel geometry and magnetic field information, and outputs key performance metrics like thrust and prediction accuracy, enabling efficient thruster design and performance analysis. >
Additionally, Professor Choe mentioned, “The CubeSat Hall thruster, developed using the AI technique in collaboration with our lab startup—Cosmo Bee, an electric propulsion company—will be tested in orbit this November aboard the K-HERO 3U (30 x 10 x 10 cm) CubeSat, scheduled for launch on the fourth flight of the KSLV-2 Nuri rocket.”
This research was published online in Advanced Intelligent Systems on December 25, 2024 with PhD candidate Jaehong Park as the first author and was selected as the journal’s cover article, highlighting its innovation.
< Figure 3. Image of the 150 W low-power Hall thruster for small and micro satellites, developed in collaboration with Cosmo Bee and the KAIST team. The thruster will be tested in orbit on the K-HERO CubeSat during the KSLV-2 Nuri rocket’s fourth launch in Q4 2025. >
This research was supported by the National Research Foundation of Korea’s Space Pioneer Program (200mN High Thrust Electric Propulsion System Development).
(Paper Title: Predicting Performance of Hall Effect Ion Source Using Machine Learning, DOI: https://doi.org/10.1002/aisy.202400555 )
< Figure 4. Graphs of the predicted thrust and discharge current of KAIST’s 700 W Hall thruster using the AI model (HallNN). The left image shows the Hall thruster operating in KAIST Electric Propulsion Laboratory’s vacuum chamber, while the center and right graphs present the prediction results for thrust and discharge current based on anode mass flow rate. The red lines represent AI predictions, and the blue dots represent experimental results, with a prediction error of less than 5%. >
2025.02.03 View 5889 -
 KAIST Team Develops an Insect-Mimicking Semiconductor to Detect Motion
The recent development of an “intelligent sensor” semiconductor that mimics the optic nerve of insects while operating at ultra-high speeds and low power offers extensive expandability into various innovative technologies. This technology is expected to be applied to various fields including transportation, safety, and security systems, contributing to both industry and society.
On February 19, a KAIST research team led by Professor Kyung Min Kim from the Department of Materials Science and Engineering (DMSE) announced the successful developed an intelligent motion detector by merging various memristor* devices to mimic the visual intelligence** of the optic nerve of insects.
*Memristor: a “memory resistor” whose state of resistance changes depending on the input signal
**Visual intelligence: the ability to interpret visual information and perform calculations within the optic nerve
With the recent advances in AI technology, vision systems are being improved by utilizing AI in various tasks such as image recognition, object detection, and motion analysis. However, existing vision systems typically recognize objects and their behaviour from the received image signals using complex algorithms. This method requires a significant amount of data traffic and higher power consumption, making it difficult to apply in mobile or IoT devices.
Meanwhile, insects are known to be able to effectively process visual information through an optic nerve circuit called the elementary motion detector, allowing them to detect objects and recognize their motion at an advanced level. However, mimicking this pathway using conventional silicon integrated circuit (CMOS) technology requires complex circuits, and its implementation into actual devices has thus been limited.
< Figure 1. Working principle of a biological elementary motion detection system. >
Professor Kyung Min Kim’s research team developed an intelligent motion detecting sensor that operates at a high level of efficiency and ultra-high speeds. The device has a simple structure consisting of only two types of memristors and a resistor developed by the team. The two different memristors each carry out a signal delay function and a signal integration and ignition function, respectively. Through them, the team could directly mimic the optic nerve of insects to analyze object movement.
< Figure 2. (Left) Optical image of the M-EMD device in the left panel (scale bar 200 μm) and SEM image of the device in the right panel (scale bar: 20 μm). (Middle) Responses of the M-EMD in positive direction. (Right) Responses of the M-EMD in negative direction. >
To demonstrate its potential for practical applications, the research team used the newly developed motion detector to design a neuromorphic computing system that can predict the path of a vehicle. The results showed that the device used 92.9% less energy compared to existing technology and predicted motion with more accuracy.
< Figure 3. Neuromorphic computing system configuration based on motion recognition devices >
Professor Kim said, “Insects make use of their very simple visual intelligence systems to detect the motion of objects at a surprising high speed. This research is significant in that we could mimic the functions of a nerve using a memristor device.” He added, “Edge AI devices, such as AI-topped mobile phones, are becoming increasingly important. This research can contribute to the integration of efficient vision systems for motion recognition, so we expect it to be applied to various fields such as autonomous vehicles, vehicle transportation systems, robotics, and machine vision.”
This research, conducted by co-first authors Hanchan Song and Min Gu Lee, both Ph.D. candidates at KAIST DMSE, was published in the online issue of Advanced Materials on January 29.
This research was supported by the Mid-Sized Research Project by the National Research Foundation of Korea, the Next-Generation Intelligent Semiconductor Technology Development Project, the PIM Artificial Intelligence Semiconductor Core Technology Development Project, the National Nano Fab Center, and the Leap Research Project by KAIST.
2024.02.29 View 7916
KAIST Team Develops an Insect-Mimicking Semiconductor to Detect Motion
The recent development of an “intelligent sensor” semiconductor that mimics the optic nerve of insects while operating at ultra-high speeds and low power offers extensive expandability into various innovative technologies. This technology is expected to be applied to various fields including transportation, safety, and security systems, contributing to both industry and society.
On February 19, a KAIST research team led by Professor Kyung Min Kim from the Department of Materials Science and Engineering (DMSE) announced the successful developed an intelligent motion detector by merging various memristor* devices to mimic the visual intelligence** of the optic nerve of insects.
*Memristor: a “memory resistor” whose state of resistance changes depending on the input signal
**Visual intelligence: the ability to interpret visual information and perform calculations within the optic nerve
With the recent advances in AI technology, vision systems are being improved by utilizing AI in various tasks such as image recognition, object detection, and motion analysis. However, existing vision systems typically recognize objects and their behaviour from the received image signals using complex algorithms. This method requires a significant amount of data traffic and higher power consumption, making it difficult to apply in mobile or IoT devices.
Meanwhile, insects are known to be able to effectively process visual information through an optic nerve circuit called the elementary motion detector, allowing them to detect objects and recognize their motion at an advanced level. However, mimicking this pathway using conventional silicon integrated circuit (CMOS) technology requires complex circuits, and its implementation into actual devices has thus been limited.
< Figure 1. Working principle of a biological elementary motion detection system. >
Professor Kyung Min Kim’s research team developed an intelligent motion detecting sensor that operates at a high level of efficiency and ultra-high speeds. The device has a simple structure consisting of only two types of memristors and a resistor developed by the team. The two different memristors each carry out a signal delay function and a signal integration and ignition function, respectively. Through them, the team could directly mimic the optic nerve of insects to analyze object movement.
< Figure 2. (Left) Optical image of the M-EMD device in the left panel (scale bar 200 μm) and SEM image of the device in the right panel (scale bar: 20 μm). (Middle) Responses of the M-EMD in positive direction. (Right) Responses of the M-EMD in negative direction. >
To demonstrate its potential for practical applications, the research team used the newly developed motion detector to design a neuromorphic computing system that can predict the path of a vehicle. The results showed that the device used 92.9% less energy compared to existing technology and predicted motion with more accuracy.
< Figure 3. Neuromorphic computing system configuration based on motion recognition devices >
Professor Kim said, “Insects make use of their very simple visual intelligence systems to detect the motion of objects at a surprising high speed. This research is significant in that we could mimic the functions of a nerve using a memristor device.” He added, “Edge AI devices, such as AI-topped mobile phones, are becoming increasingly important. This research can contribute to the integration of efficient vision systems for motion recognition, so we expect it to be applied to various fields such as autonomous vehicles, vehicle transportation systems, robotics, and machine vision.”
This research, conducted by co-first authors Hanchan Song and Min Gu Lee, both Ph.D. candidates at KAIST DMSE, was published in the online issue of Advanced Materials on January 29.
This research was supported by the Mid-Sized Research Project by the National Research Foundation of Korea, the Next-Generation Intelligent Semiconductor Technology Development Project, the PIM Artificial Intelligence Semiconductor Core Technology Development Project, the National Nano Fab Center, and the Leap Research Project by KAIST.
2024.02.29 View 7916 -
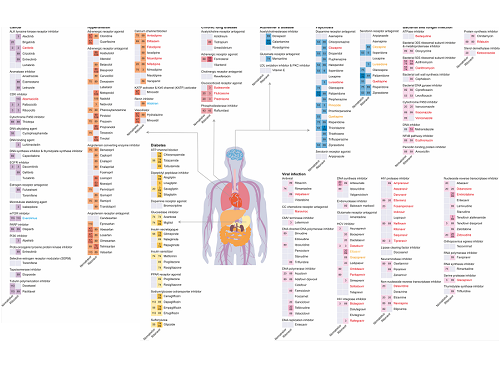 KAIST leads AI-based analysis on drug-drug interactions involving Paxlovid
KAIST (President Kwang Hyung Lee) announced on the 16th that an advanced AI-based drug interaction prediction technology developed by the Distinguished Professor Sang Yup Lee's research team in the Department of Biochemical Engineering that analyzed the interaction between the PaxlovidTM ingredients that are used as COVID-19 treatment and other prescription drugs was published as a thesis. This paper was published in the online edition of 「Proceedings of the National Academy of Sciences of America」 (PNAS), an internationally renowned academic journal, on the 13th of March.
* Thesis Title: Computational prediction of interactions between Paxlovid and prescription drugs (Authored by Yeji Kim (KAIST, co-first author), Jae Yong Ryu (Duksung Women's University, co-first author), Hyun Uk Kim (KAIST, co-first author), and Sang Yup Lee (KAIST, corresponding author))
In this study, the research team developed DeepDDI2, an advanced version of DeepDDI, an AI-based drug interaction prediction model they developed in 2018. DeepDDI2 is able to compute for and process a total of 113 drug-drug interaction (DDI) types, more than the 86 DDI types covered by the existing DeepDDI.
The research team used DeepDDI2 to predict possible interactions between the ingredients (ritonavir, nirmatrelvir) of Paxlovid*, a COVID-19 treatment, and other prescription drugs. The research team said that while among COVID-19 patients, high-risk patients with chronic diseases such as high blood pressure and diabetes are likely to be taking other drugs, drug-drug interactions and adverse drug reactions for Paxlovid have not been sufficiently analyzed, yet. This study was pursued in light of seeing how continued usage of the drug may lead to serious and unwanted complications.
* Paxlovid: Paxlovid is a COVID-19 treatment developed by Pfizer, an American pharmaceutical company, and received emergency use approval (EUA) from the US Food and Drug Administration (FDA) in December 2021.
The research team used DeepDDI2 to predict how Paxrovid's components, ritonavir and nirmatrelvir, would interact with 2,248 prescription drugs. As a result of the prediction, ritonavir was predicted to interact with 1,403 prescription drugs and nirmatrelvir with 673 drugs.
Using the prediction results, the research team proposed alternative drugs with the same mechanism but low drug interaction potential for prescription drugs with high adverse drug events (ADEs). Accordingly, 124 alternative drugs that could reduce the possible adverse DDI with ritonavir and 239 alternative drugs for nirmatrelvir were identified.
Through this research achievement, it became possible to use an deep learning technology to accurately predict drug-drug interactions (DDIs), and this is expected to play an important role in the digital healthcare, precision medicine and pharmaceutical industries by providing useful information in the process of developing new drugs and making prescriptions.
Distinguished Professor Sang Yup Lee said, "The results of this study are meaningful at times like when we would have to resort to using drugs that are developed in a hurry in the face of an urgent situations like the COVID-19 pandemic, that it is now possible to identify and take necessary actions against adverse drug reactions caused by drug-drug interactions very quickly.”
This research was carried out with the support of the KAIST New-Deal Project for COVID-19 Science and Technology and the Bio·Medical Technology Development Project supported by the Ministry of Science and ICT.
Figure 1. Results of drug interaction prediction between Paxlovid ingredients and representative approved drugs using DeepDDI2
2023.03.16 View 9501
KAIST leads AI-based analysis on drug-drug interactions involving Paxlovid
KAIST (President Kwang Hyung Lee) announced on the 16th that an advanced AI-based drug interaction prediction technology developed by the Distinguished Professor Sang Yup Lee's research team in the Department of Biochemical Engineering that analyzed the interaction between the PaxlovidTM ingredients that are used as COVID-19 treatment and other prescription drugs was published as a thesis. This paper was published in the online edition of 「Proceedings of the National Academy of Sciences of America」 (PNAS), an internationally renowned academic journal, on the 13th of March.
* Thesis Title: Computational prediction of interactions between Paxlovid and prescription drugs (Authored by Yeji Kim (KAIST, co-first author), Jae Yong Ryu (Duksung Women's University, co-first author), Hyun Uk Kim (KAIST, co-first author), and Sang Yup Lee (KAIST, corresponding author))
In this study, the research team developed DeepDDI2, an advanced version of DeepDDI, an AI-based drug interaction prediction model they developed in 2018. DeepDDI2 is able to compute for and process a total of 113 drug-drug interaction (DDI) types, more than the 86 DDI types covered by the existing DeepDDI.
The research team used DeepDDI2 to predict possible interactions between the ingredients (ritonavir, nirmatrelvir) of Paxlovid*, a COVID-19 treatment, and other prescription drugs. The research team said that while among COVID-19 patients, high-risk patients with chronic diseases such as high blood pressure and diabetes are likely to be taking other drugs, drug-drug interactions and adverse drug reactions for Paxlovid have not been sufficiently analyzed, yet. This study was pursued in light of seeing how continued usage of the drug may lead to serious and unwanted complications.
* Paxlovid: Paxlovid is a COVID-19 treatment developed by Pfizer, an American pharmaceutical company, and received emergency use approval (EUA) from the US Food and Drug Administration (FDA) in December 2021.
The research team used DeepDDI2 to predict how Paxrovid's components, ritonavir and nirmatrelvir, would interact with 2,248 prescription drugs. As a result of the prediction, ritonavir was predicted to interact with 1,403 prescription drugs and nirmatrelvir with 673 drugs.
Using the prediction results, the research team proposed alternative drugs with the same mechanism but low drug interaction potential for prescription drugs with high adverse drug events (ADEs). Accordingly, 124 alternative drugs that could reduce the possible adverse DDI with ritonavir and 239 alternative drugs for nirmatrelvir were identified.
Through this research achievement, it became possible to use an deep learning technology to accurately predict drug-drug interactions (DDIs), and this is expected to play an important role in the digital healthcare, precision medicine and pharmaceutical industries by providing useful information in the process of developing new drugs and making prescriptions.
Distinguished Professor Sang Yup Lee said, "The results of this study are meaningful at times like when we would have to resort to using drugs that are developed in a hurry in the face of an urgent situations like the COVID-19 pandemic, that it is now possible to identify and take necessary actions against adverse drug reactions caused by drug-drug interactions very quickly.”
This research was carried out with the support of the KAIST New-Deal Project for COVID-19 Science and Technology and the Bio·Medical Technology Development Project supported by the Ministry of Science and ICT.
Figure 1. Results of drug interaction prediction between Paxlovid ingredients and representative approved drugs using DeepDDI2
2023.03.16 View 9501 -
 Scientists re-writes FDA-recommended equation to improve estimation of drug-drug interaction
Drugs absorbed into the body are metabolized and thus removed by enzymes from several organs like the liver. How fast a drug is cleared out of the system can be affected by other drugs that are taken together because added substance can increase the amount of enzyme secretion in the body. This dramatically decreases the concentration of a drug, reducing its efficacy, often leading to the failure of having any effect at all. Therefore, accurately predicting the clearance rate in the presence of drug-drug interaction* is critical in the process of drug prescription and development of a new drug in order to ensure its efficacy and/or to avoid unwanted side-effects.
*Drug-drug interaction: In terms of metabolism, drug-drug interaction is a phenomenon in which one drug changes the metabolism of another drug to promote or inhibit its excretion from the body when two or more drugs are taken together. As a result, it increases the toxicity of medicines or causes loss of efficacy.
Since it is practically impossible to evaluate all interactions between new drug candidates and all marketed drugs during the development process, the FDA recommends indirect evaluation of drug interactions using a formula suggested in their guidance, first published in 1997, revised in January of 2020, in order to evaluate drug interactions and minimize side effects of having to use more than one type of drugs at once.
The formula relies on the 110-year-old Michaelis-Menten (MM) model, which has a fundamental limit of making a very broad and groundless assumption on the part of the presence of the enzymes that metabolizes the drug. While MM equation has been one of the most widely known equations in biochemistry used in more than 220,000 published papers, the MM equation is accurate only when the concentration of the enzyme that metabolizes the drug is almost non-existent, causing the accuracy of the equation highly unsatisfactory – only 38 percent of the predictions had less than two-fold errors.
“To make up for the gap, researcher resorted to plugging in scientifically unjustified constants into the equation,” Professor Jung-woo Chae of Chungnam National University College of Pharmacy said. “This is comparable to having to have the epicyclic orbits introduced to explain the motion of the planets back in the days in order to explain the now-defunct Ptolemaic theory, because it was 'THE' theory back then.”
< (From left) Ph.D. student Yun Min Song (KAIST, co-first authors), Professor Sang Kyum Kim (Chungnam National University, co-corresponding author), Jae Kyoung Kim, CI (KAIST, co-corresponding author), Professor Jung-woo Chae (Chungnam National University, co-corresponding author), Ph.D. students Quyen Thi Tran and Ngoc-Anh Thi Vu (Chungnam National University, co-first authors) >
A joint research team composed of mathematicians from the Biomedical Mathematics Group within the Institute for Basic Science (IBS) and the Korea Advanced Institute of Science and Technology (KAIST) and pharmacological scientists from the Chungnam National University reported that they identified the major causes of the FDA-recommended equation’s inaccuracies and presented a solution.
When estimating the gut bioavailability (Fg), which is the key parameter of the equation, the fraction absorbed from the gut lumen (Fa) is usually assumed to be 1. However, many experiments have shown that Fa is less than 1, obviously since it can’t be expected that all of the orally taken drugs to be completely absorbed by the intestines. To solve this problem, the research team used an “estimated Fa” value based on factors such as the drug’s transit time, intestine radius, and permeability values and used it to re-calculate Fg.
Also, taking a different approach from the MM equation, the team used an alternative model they derived in a previous study back in 2020, which can more accurately predict the drug metabolism rate regardless of the enzyme concentration. Combining these changes, the modified equation with re-calculated Fg had a dramatically increased accuracy of the resulting estimate. The existing FDA formula predicted drug interactions within a 2-fold margin of error at the rate of 38%, whereas the accuracy rate of the revised formula reached 80%.
“Such drastic improvement in drug-drug interaction prediction accuracy is expected to make great contribution to increasing the success rate of new drug development and drug efficacy in clinical trials. As the results of this study were published in one of the top clinical pharmacology journal, it is expected that the FDA guidance will be revised according to the results of this study.” said Professor Sang Kyum Kim from Chungnam National University College of Pharmacy.
Furthermore, this study highlights the importance of collaborative research between research groups in vastly different disciplines, in a field that is as dynamic as drug interactions.
“Thanks to the collaborative research between mathematics and pharmacy, we were able to recify the formula that we have accepted to be the right answer for so long to finally grasp on the leads toward healthier life for mankind.,” said Professor Jae Kyung Kim. He continued, “I hope seeing a ‘K-formula’ entered into the US FDA guidance one day.”
The results of this study were published in the online edition of Clinical Pharmacology and Therapeutics (IF 7.051), an authoritative journal in the field of clinical pharmacology, on December 15, 2022 (Korean time).
Thesis Title: Beyond the Michaelis-Menten: Accurate Prediction of Drug Interactions through Cytochrome P450 3A4 Induction (doi: 10.1002/cpt.2824)
< Figure 1. The formula proposed by the FDA guidance for predicting drug-drug interactions (top) and the formula newly derived by the researchers (bottom). AUCR (the ratio of substrate area under the plasma concentration-time curve) represents the rate of change in drug concentration due to drug interactions. The research team more than doubled the accuracy of drug interaction prediction compared to the existing formula. >
< Figure 2. Existing FDA formulas tend to underestimate the extent of drug-drug interactions (gray dots) than the actual measured values. On the other hand, the newly derived equation (red dot) has a prediction rate that is within the error range of 2 times (0.5 to 2 times) of the measured value, and is more than twice as high as the existing equation. The solid line in the figure represents the predicted value that matches the measured value. The dotted line represents the predicted value with an error of 0.5 to 2 times. >
For further information or to request media assistance, please contact Jae Kyoung Kim at Biomedical Mathematics Group, Institute for Basic Science (IBS) (jaekkim@ibs.re.kr) or William I. Suh at the IBS Communications Team (willisuh@ibs.re.kr).
- About the Institute for Basic Science (IBS)
IBS was founded in 2011 by the government of the Republic of Korea with the sole purpose of driving forward the development of basic science in South Korea. IBS has 4 research institutes and 33 research centers as of January 2023. There are eleven physics, three mathematics, five chemistry, nine life science, two earth science, and three interdisciplinary research centers.
2023.01.18 View 14461
Scientists re-writes FDA-recommended equation to improve estimation of drug-drug interaction
Drugs absorbed into the body are metabolized and thus removed by enzymes from several organs like the liver. How fast a drug is cleared out of the system can be affected by other drugs that are taken together because added substance can increase the amount of enzyme secretion in the body. This dramatically decreases the concentration of a drug, reducing its efficacy, often leading to the failure of having any effect at all. Therefore, accurately predicting the clearance rate in the presence of drug-drug interaction* is critical in the process of drug prescription and development of a new drug in order to ensure its efficacy and/or to avoid unwanted side-effects.
*Drug-drug interaction: In terms of metabolism, drug-drug interaction is a phenomenon in which one drug changes the metabolism of another drug to promote or inhibit its excretion from the body when two or more drugs are taken together. As a result, it increases the toxicity of medicines or causes loss of efficacy.
Since it is practically impossible to evaluate all interactions between new drug candidates and all marketed drugs during the development process, the FDA recommends indirect evaluation of drug interactions using a formula suggested in their guidance, first published in 1997, revised in January of 2020, in order to evaluate drug interactions and minimize side effects of having to use more than one type of drugs at once.
The formula relies on the 110-year-old Michaelis-Menten (MM) model, which has a fundamental limit of making a very broad and groundless assumption on the part of the presence of the enzymes that metabolizes the drug. While MM equation has been one of the most widely known equations in biochemistry used in more than 220,000 published papers, the MM equation is accurate only when the concentration of the enzyme that metabolizes the drug is almost non-existent, causing the accuracy of the equation highly unsatisfactory – only 38 percent of the predictions had less than two-fold errors.
“To make up for the gap, researcher resorted to plugging in scientifically unjustified constants into the equation,” Professor Jung-woo Chae of Chungnam National University College of Pharmacy said. “This is comparable to having to have the epicyclic orbits introduced to explain the motion of the planets back in the days in order to explain the now-defunct Ptolemaic theory, because it was 'THE' theory back then.”
< (From left) Ph.D. student Yun Min Song (KAIST, co-first authors), Professor Sang Kyum Kim (Chungnam National University, co-corresponding author), Jae Kyoung Kim, CI (KAIST, co-corresponding author), Professor Jung-woo Chae (Chungnam National University, co-corresponding author), Ph.D. students Quyen Thi Tran and Ngoc-Anh Thi Vu (Chungnam National University, co-first authors) >
A joint research team composed of mathematicians from the Biomedical Mathematics Group within the Institute for Basic Science (IBS) and the Korea Advanced Institute of Science and Technology (KAIST) and pharmacological scientists from the Chungnam National University reported that they identified the major causes of the FDA-recommended equation’s inaccuracies and presented a solution.
When estimating the gut bioavailability (Fg), which is the key parameter of the equation, the fraction absorbed from the gut lumen (Fa) is usually assumed to be 1. However, many experiments have shown that Fa is less than 1, obviously since it can’t be expected that all of the orally taken drugs to be completely absorbed by the intestines. To solve this problem, the research team used an “estimated Fa” value based on factors such as the drug’s transit time, intestine radius, and permeability values and used it to re-calculate Fg.
Also, taking a different approach from the MM equation, the team used an alternative model they derived in a previous study back in 2020, which can more accurately predict the drug metabolism rate regardless of the enzyme concentration. Combining these changes, the modified equation with re-calculated Fg had a dramatically increased accuracy of the resulting estimate. The existing FDA formula predicted drug interactions within a 2-fold margin of error at the rate of 38%, whereas the accuracy rate of the revised formula reached 80%.
“Such drastic improvement in drug-drug interaction prediction accuracy is expected to make great contribution to increasing the success rate of new drug development and drug efficacy in clinical trials. As the results of this study were published in one of the top clinical pharmacology journal, it is expected that the FDA guidance will be revised according to the results of this study.” said Professor Sang Kyum Kim from Chungnam National University College of Pharmacy.
Furthermore, this study highlights the importance of collaborative research between research groups in vastly different disciplines, in a field that is as dynamic as drug interactions.
“Thanks to the collaborative research between mathematics and pharmacy, we were able to recify the formula that we have accepted to be the right answer for so long to finally grasp on the leads toward healthier life for mankind.,” said Professor Jae Kyung Kim. He continued, “I hope seeing a ‘K-formula’ entered into the US FDA guidance one day.”
The results of this study were published in the online edition of Clinical Pharmacology and Therapeutics (IF 7.051), an authoritative journal in the field of clinical pharmacology, on December 15, 2022 (Korean time).
Thesis Title: Beyond the Michaelis-Menten: Accurate Prediction of Drug Interactions through Cytochrome P450 3A4 Induction (doi: 10.1002/cpt.2824)
< Figure 1. The formula proposed by the FDA guidance for predicting drug-drug interactions (top) and the formula newly derived by the researchers (bottom). AUCR (the ratio of substrate area under the plasma concentration-time curve) represents the rate of change in drug concentration due to drug interactions. The research team more than doubled the accuracy of drug interaction prediction compared to the existing formula. >
< Figure 2. Existing FDA formulas tend to underestimate the extent of drug-drug interactions (gray dots) than the actual measured values. On the other hand, the newly derived equation (red dot) has a prediction rate that is within the error range of 2 times (0.5 to 2 times) of the measured value, and is more than twice as high as the existing equation. The solid line in the figure represents the predicted value that matches the measured value. The dotted line represents the predicted value with an error of 0.5 to 2 times. >
For further information or to request media assistance, please contact Jae Kyoung Kim at Biomedical Mathematics Group, Institute for Basic Science (IBS) (jaekkim@ibs.re.kr) or William I. Suh at the IBS Communications Team (willisuh@ibs.re.kr).
- About the Institute for Basic Science (IBS)
IBS was founded in 2011 by the government of the Republic of Korea with the sole purpose of driving forward the development of basic science in South Korea. IBS has 4 research institutes and 33 research centers as of January 2023. There are eleven physics, three mathematics, five chemistry, nine life science, two earth science, and three interdisciplinary research centers.
2023.01.18 View 14461 -
 Bioengineers develop a new strategy for accurate prediction of cellular metabolic fluxes
A team of pioneering South Korean scientists has developed a new strategy for accurately predicting cellular metabolic fluxes under various genotypic and environmental conditions. This groundbreaking research is published in the journal Proceedings of the National Academy of Sciences of the USA (PNAS) on August 2, 2010.
To understand cellular metabolism and predict its metabolic capability at systems-level, systems biological analysis by modeling and simulation of metabolic network plays an important role. The team from the Korea Advanced Institute of Science and Technology (KAIST), led by Distinguished Professor Sang Yup Lee, focused their research on the development of a new strategy for more accurate prediction of cellular metabolism.
“For strain improvement, biologists have made every effort to understand the global picture of biological systems and investigate the changes of all metabolic fluxes of the system under changing genotypic and environmental conditions,” said Lee. The accumulation of omics data, including genome, transcriptome, proteome, metabolome, and fluxome, provides an opportunity to understand the cellular physiology and metabolic characteristics at systems-level. With the availability of the fully annotated genome sequence, the genome-scale in silico (means “performed on computer or via computer simulation.”) metabolic models for a number of organisms have been successfully developed to improve our understanding on these biological systems. With these advances, the development of new simulation methods to analyze and integrate systematically large amounts of biological data and predict cellular metabolic capability for systems biological analysis is important.
Information used to reconstruct the genome-scale in silico cell is not yet complete, which can make the simulation results different from the physiological performances of the real cell. Thus, additional information and procedures, such as providing additional constraints (constraint: a term to exclude incorrect metabolic fluxes by restricting the solution space of in silico cell) to the model, are often incorporated to improve the accuracy of the in silico cell.
By employing information generated from the genome sequence and annotation, the KAIST team developed a new set of constraints, called Grouping Reaction (GR) constraints, to accurately predict metabolic fluxes. Based on the genomic information, functionally related reactions were organized into different groups. These groups were considered for the generation of GR constraints, as condition- and objective function- independent constraints. Since the method developed in this study does not require complex information but only the genome sequence and annotation, this strategy can be applied to any organism with a completely annotated genome sequence.
“As we become increasingly concerned with environmental problems and the limits of fossil resources, bio-based production of chemicals from renewable biomass has been receiving great attention. Systems biological analysis by modeling and simulation of biological systems, to understand cellular metabolism and identify the targets for the strain improvement, has provided a new paradigm for developing successful bioprocesses,” concluded Lee. This new strategy for predicting cellular metabolism is expected to contribute to more accurate determination of cellular metabolic characteristics, and consequently to the development of metabolic engineering strategies for the efficient production of important industrial products and identification of new drug targets in pathogens.”
2010.08.05 View 16175
Bioengineers develop a new strategy for accurate prediction of cellular metabolic fluxes
A team of pioneering South Korean scientists has developed a new strategy for accurately predicting cellular metabolic fluxes under various genotypic and environmental conditions. This groundbreaking research is published in the journal Proceedings of the National Academy of Sciences of the USA (PNAS) on August 2, 2010.
To understand cellular metabolism and predict its metabolic capability at systems-level, systems biological analysis by modeling and simulation of metabolic network plays an important role. The team from the Korea Advanced Institute of Science and Technology (KAIST), led by Distinguished Professor Sang Yup Lee, focused their research on the development of a new strategy for more accurate prediction of cellular metabolism.
“For strain improvement, biologists have made every effort to understand the global picture of biological systems and investigate the changes of all metabolic fluxes of the system under changing genotypic and environmental conditions,” said Lee. The accumulation of omics data, including genome, transcriptome, proteome, metabolome, and fluxome, provides an opportunity to understand the cellular physiology and metabolic characteristics at systems-level. With the availability of the fully annotated genome sequence, the genome-scale in silico (means “performed on computer or via computer simulation.”) metabolic models for a number of organisms have been successfully developed to improve our understanding on these biological systems. With these advances, the development of new simulation methods to analyze and integrate systematically large amounts of biological data and predict cellular metabolic capability for systems biological analysis is important.
Information used to reconstruct the genome-scale in silico cell is not yet complete, which can make the simulation results different from the physiological performances of the real cell. Thus, additional information and procedures, such as providing additional constraints (constraint: a term to exclude incorrect metabolic fluxes by restricting the solution space of in silico cell) to the model, are often incorporated to improve the accuracy of the in silico cell.
By employing information generated from the genome sequence and annotation, the KAIST team developed a new set of constraints, called Grouping Reaction (GR) constraints, to accurately predict metabolic fluxes. Based on the genomic information, functionally related reactions were organized into different groups. These groups were considered for the generation of GR constraints, as condition- and objective function- independent constraints. Since the method developed in this study does not require complex information but only the genome sequence and annotation, this strategy can be applied to any organism with a completely annotated genome sequence.
“As we become increasingly concerned with environmental problems and the limits of fossil resources, bio-based production of chemicals from renewable biomass has been receiving great attention. Systems biological analysis by modeling and simulation of biological systems, to understand cellular metabolism and identify the targets for the strain improvement, has provided a new paradigm for developing successful bioprocesses,” concluded Lee. This new strategy for predicting cellular metabolism is expected to contribute to more accurate determination of cellular metabolic characteristics, and consequently to the development of metabolic engineering strategies for the efficient production of important industrial products and identification of new drug targets in pathogens.”
2010.08.05 View 16175 -
 Ju-pyeong Lee won the Best Paper Award from IEEE RTAS
Ju-pyeong Lee, doctoral student of the Dept. of Electrical Engineering of KAIST, received the Best Paper Award from the 11th Institute of Electrical and Electronics Engineers Real-Time and Embedded Technology and Applications Symposium (IEEE RTAS) sponsored by IEEE TC on Real Time System and supported from the U.S. National Science Foundation.
He is in the Computer Engineering laboratory, and won the honor by his research of technique of Delayed Locking Technique for Improving Real-Time Performance of Embedded Linux by Prediction of Timer Interrupt. His paper was selected to be the best because of its practicality. His research purposed the technique that can dramatically improve real time problem, which was indicated to be the big problem of Linux. Moreover, he presented the way to easily materialize this technique in the practical system.
Best Paper Award is the prize awarded by IEEE Computer Society in the recognition of outstanding achievement in the field of real time system and embedded technology.
IEEE RTAS is a symposium held annually by IEEE. In this year, the 11th symposium was held from March 7 to March 10, for four days, in San Francisco, United States. The purpose of this year symposium was to seek papers describing significant contributions both to state of the art and state of the practice in the broad field of embedded and open real-time computing, control, and communication. Therefore, it especially focused on online real-time and embedded applications ranging from industrial embedded applications such as aeronautics and automotive systems to open multimedia, telecommunication and mobile computing systems.
Approximately 200 related erudite from almost 20 countries including United States, England, France, Germany, Italy, and Sweden participated in this symposium. Total number of papers submitted to IEEE RTAS was 158, while only 53 of them were selected.
by Hye-jung Won / Staff ReporterApril, 2005 / The KAIST Herald
2005.04.12 View 20397
Ju-pyeong Lee won the Best Paper Award from IEEE RTAS
Ju-pyeong Lee, doctoral student of the Dept. of Electrical Engineering of KAIST, received the Best Paper Award from the 11th Institute of Electrical and Electronics Engineers Real-Time and Embedded Technology and Applications Symposium (IEEE RTAS) sponsored by IEEE TC on Real Time System and supported from the U.S. National Science Foundation.
He is in the Computer Engineering laboratory, and won the honor by his research of technique of Delayed Locking Technique for Improving Real-Time Performance of Embedded Linux by Prediction of Timer Interrupt. His paper was selected to be the best because of its practicality. His research purposed the technique that can dramatically improve real time problem, which was indicated to be the big problem of Linux. Moreover, he presented the way to easily materialize this technique in the practical system.
Best Paper Award is the prize awarded by IEEE Computer Society in the recognition of outstanding achievement in the field of real time system and embedded technology.
IEEE RTAS is a symposium held annually by IEEE. In this year, the 11th symposium was held from March 7 to March 10, for four days, in San Francisco, United States. The purpose of this year symposium was to seek papers describing significant contributions both to state of the art and state of the practice in the broad field of embedded and open real-time computing, control, and communication. Therefore, it especially focused on online real-time and embedded applications ranging from industrial embedded applications such as aeronautics and automotive systems to open multimedia, telecommunication and mobile computing systems.
Approximately 200 related erudite from almost 20 countries including United States, England, France, Germany, Italy, and Sweden participated in this symposium. Total number of papers submitted to IEEE RTAS was 158, while only 53 of them were selected.
by Hye-jung Won / Staff ReporterApril, 2005 / The KAIST Herald
2005.04.12 View 20397