research
A research team from KAIST developed a computational framework that enables the reconstruction of a comprehensive computational model of human metabolism, which allows for an accurate prediction of personal metabolic features (or phenotypes).
Understanding personal metabolic phenotypes allows us to design effective therapeutic strategies for various chronic and infectious diseases. A human computational model called the genome-scale metabolic model (GEM) contains information on thousands of metabolic genes and their corresponding reactions and metabolites, and has played an important role in predicting metabolic phenotypes. Although several versions of human GEMs have been released, they had room for further development, especially as to incorporating biological information coming from a human genetics mechanism called “alternative splicing.” Alternative splicing is a genetic mechanism that allows a gene to give rise to multiple reactions, and is strongly associated with pathology.
To tackle this problem, Jae Yong Ryu (a Ph.D. student), Dr. Hyun Uk Kim (Research Fellow), and Distinguished Professor Sang Yup Lee, all from the Department of Chemical and Biomolecular Engineering at KAIST, developed a computational framework that systematically generates metabolic reactions, and adds them to the human GEM. The resulting human GEM was demonstrated to accurately predict metabolic phenotypes under varied environmental conditions. The research results were published online in Proceedings of the National Academy of Sciences (PNAS) on October 24, 2017, under the title “Framework and resource for more than 11,000 gene-transcript-protein-reaction associations in human metabolism.”
The research team first updated the biological contents of a previous version of the human GEM. The updated biological contents include metabolic genes and their corresponding metabolites and reactions. In particular, metabolic reactions catalyzed by already-known protein isoforms were additionally incorporated into the human GEM; protein isoforms are multiple variants of proteins generated from individual genes through the alternative splicing process. Each protein isoform is often responsible for the operation of a metabolic reaction. Although multiple protein isoforms generated from one gene can play different functions by having different sets of protein domains and/or subcellular localizations, such information was not properly considered in previous versions of human GEMs.
Upon the initial update of the human GEM, named Recon 2M.1, the research team subsequently implemented a computational framework that systematically generates information on Gene-Transcript-Protein-Reaction Associations (GeTPRA) in order to identify protein isoforms that were previously not identified. This framework was developed in this study. As a result of the implementation of the framework for GeTPRA, more than 11,000 GeTPRA were automatically predicted, and thoroughly validated. Additional metabolic reactions were then added to Recon 2M.1 based on the predicted GeTPRA for the previously uncharacterized protein isoforms; Recon 2M.1 was renamed Recon 2M.2 from this upgrade.
Finally, Recon 2M.2 was integrated with 446 sets of personal biological data (RNA-Seq data) in order to build patient-specific cancer models. These patient-specific cancer models were used to predict cancer metabolism activities and anticancer targets.
The development of a new version of human GEMs along with the computational framework for GeTPRA is expected to boost studies in fundamental human genetics and medicine. Model files of the human GEMs Recon 2M.1 and 2M.2, a full list of the GeTPRA and the source code for the computational framework to predict the GeTPRA are all available as part of the publication of this study.
Distinguished Professor Lee said, “The predicted GeTPRA from the computational framework is expected to serve as a guideline for future experiments on human genetics and biochemistry, whereas the resulting Recon 2M.2 can be used to predict drug targets for various human diseases.”
This work was supported by the Technology Development Program to Solve Climate Changes on Systems Metabolic Engineering for Biorefineries (NRF-2012M1A2A2026556 and NRF-2012M1A2A2026557) from the Ministry of Science and ICT through the National Research Foundation (NRF) of Korea.

(Figure 1:A scheme of Recon 2M.1 development and its use in reconstructing personal genome-scale metabolic models (GEMs). (A) A concept of alternative splicing of human genes and its use in Gene-Transcript-Protein-Reaction Associations (GeTPRA) of Recon 2M.1. (B) A procedure of systematic refinement of the Recon 2Q. Recon 2Q is one of the previously released human GEMs. Biochemically inconsistent reactions include unbalanced, artificial, blocked, and/or redundant reactions. Iterative manual curation was conducted while validating the Recon 2M.1. (C) Reconstruction of cancer patient-specific GEMs using Recon 2M.1 for further simulation studies. In this study, personal biological data (RNA-Seq data) were obtained from The Cancer Genome Atlas (TCGA; https://cancergenome.nih.gov/ ) across the ten cancer types.
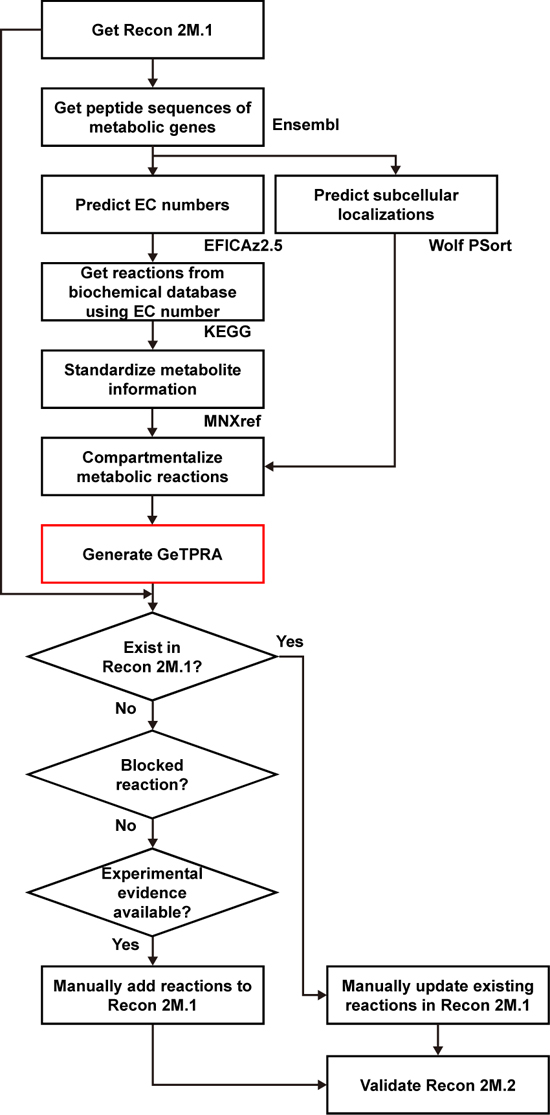
(Figure 2: Computational framework for the systematic generation of Gene-Transcript-Protein-Reaction Associations (GeTPRA; red box in the flowchart). Peptide sequences of metabolic genes defined in Recon 2M.1 were retrieved from a database called Ensembl. EC numbers and subcellular localizations of all the protein isoforms of metabolic genes in Recon 2M.1 were predicted using software programs EFICAz2.5 and Wolf PSort, respectively. Information on the newly predicted GeTPRA was systematically incorporated into the Recon 2M.1, thereby resulting in Recon 2M.2.)
-
research Engineered E. coli Using Formic Acid and CO2 As a C1-Refinery Platform Strain
(Figure: Formic acid and CO2 assimilation pathways consisting of the reconstructed THF cycle and reverse glycine cleavage reaction. This schematic diagram shows the formic acid and CO2 assimilation procedure through the pathway. Plasmids used in this study and the genetic engineering performed in this study are illustrated.) A research group at KAIST has developed an engineered E. coli strain that converts formic acid and CO2 to pyruvate and produces cellular energy from formic acid through
2018-09-18 -
research Analysis of Gas Adsorption Properties for Amorphous Porous Materials
Professor Jihan Kim from the Department of Chemical and Biomolecular Engineering at KAIST has developed a method to predict gas adsorption properties of amorphous porous materials. Metal-organic frameworks (MOFs) have large surface area and high density of pores, making them appropriate for various energy and environmental-related applications. And although most MOFs are crystalline, these structures can deform during synthesis and/or industrial processes, leading to loss in long-range order
2017-07-26