research
Cancer is characterized by abnormal metabolic processes different from those of normal cells. Therefore, cancer metabolism has been extensively studied to develop effective diagnosis and treatment strategies. Notable achievements of cancer metabolism studies include the discovery of oncometabolites* and the approval of anticancer drugs by the U.S. Food and Drug Administration (FDA) that target enzymes associated with oncometabolites. Approved anticancer drugs such as ‘Tibsovo (active ingredient: ivosidenib)’ and ‘Idhifa (active ingredient: enasidenib)’ are both used for the treatment of acute myeloid leukemia. Despite such achievements, studying cancer metabolism, especially oncometabolites, remains challenging due to time-consuming and expensive methodologies such as metabolomics. Thus, the number of confirmed oncometabolites is very small although a relatively large number of cancer-associated gene mutations have been well studied.
*Oncometabolite: A metabolite that shows pro-oncogenic function when abnormally accumulated in cancer cells. An oncometabolite is often generated as a result of gene mutations, and this accumulation promotes the growth and survival of cancer cells. Representative oncometabolites include 2-hydroxyglutarate, succinate, and fumarate.
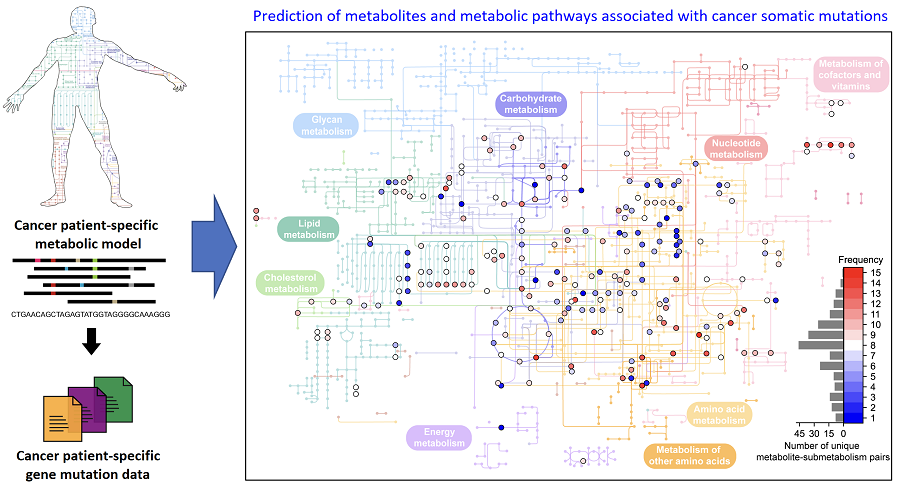
On March 18th, a KAIST research team led by Professor Hyun Uk Kim from the Department of Chemical and Biomolecular Engineering developed a computational workflow that systematically predicts metabolites and metabolic pathways associated with somatic mutations in cancer through collaboration with research teams under Prof Youngil Koh, Prof. Hongseok Yun, and Prof. Chang Wook Jeong from Seoul National University Hospital.
The research teams have successfully reconstructed patient-specific genome-scale metabolic models (GEMs)* for 1,043 cancer patients across 24 cancer types by integrating publicly available cancer patients’ transcriptome data (i.e., from international cancer genome consortiums such as PCAWG and TCGA) into a generic human GEM. The resulting patient-specific GEMs make it possible to predict each patient’s metabolic phenotypes.
*Genome-scale metabolic model (GEM): A computational model that mathematically describes all of the biochemical reactions that take place inside a cell. It allows for the prediction of the cell’s metabolic phenotypes under various genetic and/or environmental conditions.
< Figure 1. Schematic diagram of a computational methodology for predicting metabolites and metabolic pathways associated with cancer somatic mutations. of a computational methodology for predicting metabolites and metabolic pathways associated with cancer somatic mutations. >
The team developed a four-step computational workflow using the patient-specific GEMs from 1,043 cancer patients and somatic mutation data obtained from the corresponding cancer patients. This workflow begins with the calculation of the flux-sum value of each metabolite by simulating the patient-specific GEMs. The flux-sum value quantifies the intracellular importance of a metabolite. Next, the workflow identifies metabolites that appear to be significantly associated with specific gene mutations through a statistical analysis of the predicted flux-sum data and the mutation data. Finally, the workflow selects altered metabolic pathways that significantly contribute to the biosynthesis of the predicted oncometabolite candidates, ultimately generating metabolite-gene-pathway sets as an output.
The two co-first authors, Dr. GaRyoung Lee (currently a postdoctoral fellow at the Dana-Farber Cancer Institute and Harvard Medical School) and Dr. Sang Mi Lee (currently a postdoctoral fellow at Harvard Medical School) said, “The computational workflow developed can systematically predict how genetic mutations affect cellular metabolism through metabolic pathways. Importantly, it can easily be applied to different types of cancer based on the mutation and transcriptome data of cancer patient cohorts.”
Prof. Kim said, “The computational workflow and its resulting prediction outcomes will serve as the groundwork for identifying novel oncometabolites and for facilitating the development of various treatment and diagnosis strategies”.
This study, which was supported by the National Research Foundation of Korea, has been published online in Genome Biology, a representative journal in the field of biotechnology and genetics, under the title "Prediction of metabolites associated with somatic mutations in cancers by using genome‑scale metabolic models and mutation data".
-
research Genome Sequencing Unveils Mutational Impacts of Radiation on Mammalian Cells
Recent release of the waste water from Japan's Fukushima nuclear disaster stirred apprehension regarding the health implications of radiation exposure. Classified as a Group 1 carcinogen, ionizing radiation has long been associated with various cancers and genetic disorders, as evidenced by survivors and descendants of atomic bombings and the Chernobyl disaster. Despite much smaller amount, we remain consistently exposed to low levels of radiation in everyday life and medical procedures. Radi
2024-02-15 -
research A KAIST Research Team Identifies a Cancer Reversion Mechanism
Despite decades of intensive cancer research by numerous biomedical scientists, cancer still holds its place as the number one cause of death in Korea. The fundamental reason behind the limitations of current cancer treatment methods is the fact that they all aim to completely destroy cancer cells, which eventually allows the cancer cells to acquire immunity. In other words, recurrences and side-effects caused by the destruction of healthy cells are inevitable. To this end, some have suggested a
2023-06-20 -
research 'Jumping Genes' Found to Alter Human Colon Genomes, Offering Insights into Aging and Tumorigenesis
The Korea Advanced Institute of Science and Technology (KAIST) and their collaborators have conducted a groundbreaking study targeting 'jumping genes' in the entire genomes of the human large intestine. Published in Nature on May 18 2023, the research unveils the surprising activity of 'Long interspersed nuclear element-1 (L1),' a type of jumping gene previously thought to be mostly dormant in human genomes. The study shows that L1 genes can become activated and disrupt genomic functions through
2023-05-22 -
research KAIST presents a fundamental technology to remove metastatic traits from lung cancer cells
KAIST (President Kwang Hyung Lee) announced on January 30th that a research team led by Professor Kwang-Hyun Cho from the Department of Bio and Brain Engineering succeeded in using systems biology research to change the properties of carcinogenic cells in the lungs and eliminate both drug resistance and their ability to proliferate out to other areas of the body. As the incidences of cancer increase within aging populations, cancer has become the most lethal disease threatening healthy life.
2023-01-30 -
research Connecting the Dots to Find New Treatments for Breast Cancer
Systems biologists uncovered new ways of cancer cell reprogramming to treat drug-resistant cancers Scientists at KAIST believe they may have found a way to reverse an aggressive, treatment-resistant type of breast cancer into a less dangerous kind that responds well to treatment. The study involved the use of mathematical models to untangle the complex genetic and molecular interactions that occur in the two types of breast cancer, but could be extended to find ways for treating many others.
2021-12-07